Proposal of the single-atom potential confinement strategy for catalyst preparation
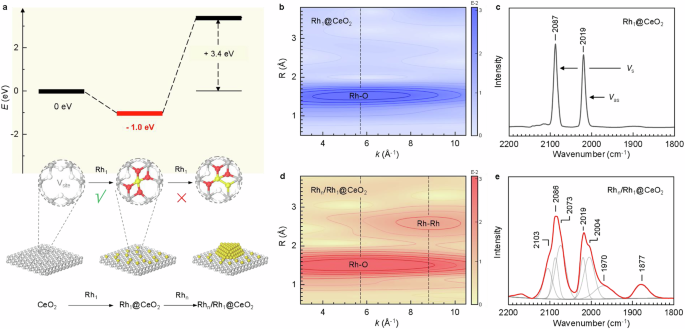
To establish the single-atom potential confinement strategy, we first employed density functional theory (DFT) calculations to investigate the energy changes (Eb) associated with stabilizing a single Rh atom at a vacancy site (Vsite) on the CeO2(111) surface, as well as the decomposition of Rh metal nanoparticles in the presence of already occupied sites (see supplementary Information for more details). The calculations showed that, for CeO2 surfaces rich in vacancies8, the adsorption of a Rh atom at a vacancy site is exothermic by 1.0 eV (Fig. 1a). This indicated that Rh nanoparticles supported on CeO2 surfaces containing vacancies can decompose readily into single Rh atoms that are stabilized at vacancy sites, in good agreement with experimental observations7,9,10,11,12,13. Interestingly, once the vacancy is occupied by a single atom, our results revealed that further dissociation of a Rh cluster to bond an additional single atom at this already-occupied site becomes energetically unfavorable, which requires substantial additional energy input (endothermic by 3.4 eV). Based on these findings, we proposed the existence of a potential confinement effect for the decomposition of Rh NPs. By leveraging this single-atom potential confinement strategy, we successfully prepared stable Rh NP catalysts on CeO2 surfaces with the pre-occupied vacancies (Fig. 1a (bottom)).

a DFT calculation for the binding energy values (E) of two individual Rh atoms (or including one oxygen atom) in sequence to the CeO2(111) surface with one Vsite (up). Structural models of the single-atom potential confinement strategy for preparing Rhn/Rh1@CeO2 (down). Yellow: Rh atoms; gray: Ce atoms; white: O atoms (the same hereinafter). b–e Rh K-edge WT EXAFS (b, d) and CO DRIFT (c, e) spectra of Rh1@CeO2 (b, c), and Rhn/Rh1@CeO2 (d, e). The bands at ~2087 and 2019 cm−1 in (c) or ~2086 and 2019 cm−1 in (e) are assigned to symmetric vibration (vs) and asymmetric vibration (vas) modes of the adsorbed CO molecules on the isolated Rh atoms on the CeO2 surfaces, respectively. The broad bands at ~1970 and 1877 cm−1 in (e) are due to the stretching vibration mode of individual adsorbed CO molecules in a bridging mode on two neighboring Rh atoms on the nanofacets of the Rh NPs. Two deconvoluted bands at ~2073 and 2004 cm−1 in (e) are assigned to vs and vas modes of the adsorbed CO molecules on the individual edge Rh atoms of the Rh NPs, respectively. The band appearing at ~2103 cm−1 (e) is due to O2−-Rh2+-CO species on the Rh NPs.
The single-atom dispersion states of Rh1@CeO2 (@ denotes anchoring, and here represents a situation of single active metal atoms bound to support surface vacancies) were verified by extended X-ray absorption fine structure (EXAFS) spectroscopy and CO diffuse reflectance infrared Fourier transform (DRIFT) spectroscopy. Figure 1b shows the Rh K-edge wavelet-transform (WT) EXAFS spectrum of Rh1@CeO2, in which only one intensity maximum could be attributed to the Rh-O scattering, and no scattering pathway due to Rh-Rh was observed (Fig. S1, Table S1), implying the isolated dispersion state of the Rh atoms. Two sharp bands in the CO DRIFT spectrum of Rh1@CeO2 (Fig. 1c) were assigned to the stretching vibration modes of two adsorbed CO molecules on one isolated Rh atom14. X-ray photoelectron spectra (XPS) revealed that the concentration of surface defect oxygen species and Ce3+ sharply decreases after the single Rh atom anchoring (Fig. S2), evidencing that the single Rh atoms have occupied the CeO2 surface Vsites. A pioneer work underlines that many oxygen vacancies can be formed in CeO2(111), such as single, double, linear, surface, and subsurface15. Therefore, it is important to clearly define the vacancy structure. The specific structure of the Rh anchoring position is determined through a comprehensive analysis of DFT calculations and the above experimental data. We compared the coordination, bond lengths, and oxidation states of Rh when anchored in different vacancy sites (Fig. S3, Table S2), and finally selected the structure that most closely matched the actual situation for subsequent calculations (Fig. S3d).
We next deposited ~2 nm Rh metallic NPs16 on Rh1@CeO2 and annealed them at 800 °C to obtain Rhn/Rh1@CeO2 (where Rhn denotes Rh NPs; Fig. S4). A new intensity maximum at ~8.8 Å−1 characteristic of the Rh-Rh bonds appeared in the WT EXAFS spectrum (Figs. 1d, S5), and DRIFT bands characteristic of metal particles17 were centered at ~1877 and 1970 cm−1 (Fig. 1e). This evidence demonstrated the survival of the Rh NPs after annealing Rhn/Rh1@CeO2 at 800 °C. Likewise, as evidenced in the WT EXAFS (Fig. 1d) and CO DRIFT (Fig. 1e) spectra, the single Rh atoms anchored at the surface Vsites were retained and co-existed with Rh NPs on Rhn/Rh1@CeO2. The results indicated that the single-atom potential confinement strategy can effectively prevent the Rh NPs from undergoing atomic dispersion at high temperatures. Owing to their affinity for oxygen, the Rh NPs were also shown to be partially covered with oxygen atoms, as evidenced by two deconvoluted bands at ~2073 and 2004 cm−1 arising from CO adsorbed on oxidized Rh atoms18, and a band at ~2103 cm−1 assigned to O2−-Rh2+-CO species on the Rh NPs19 (Fig. 1e). Such an oxygen affinity underlied the use of Rh as an effective catalyst for hydrocarbon oxidation20.
Evaluation of the single-atom potential confinement strategy in catalysis
We tested the validity of the single-atom potential confinement strategy in catalytic reaction by comparing the activity and stability of Rhn/Rh1@CeO2 with a Rhn/CeO2 reference in methane oxidation under simulated engine emission conditions21. Rhn/CeO2 was prepared by the same method as Rhn/Rh1@CeO2, except that the pristine CeO2 support and a low annealing temperature of 200 °C were used to avoid the decomposition of Rh NPs during the preparation (see supplementary Information for details). For the heating light-off curves in Fig. 2, both Rhn/Rh1@CeO2 and Rhn/CeO2 exhibit almost identical activity below ~400 °C and initiate methane oxidation at a temperature as low as 200 °C, which is comparable to the activity of well-known palladium-based catalysts3,21,22 (Tables S3, S4). Similarly, the apparent activation energy (Ea = 66 kJ mol−1) of Rhn/Rh1@CeO2 is only slightly smaller than that (Ea = 68 kJ mol−1) of Rhn/CeO223 (Fig. S6), indicating that the Rh NPs, rather than the single Rh atoms, are the active sites in Rhn/Rh1@CeO223. The conclusion was also corroborated by extremely low activity of Rh1@CeO2 and CeO2 under the identical conditions (Fig. 2a).

a, b Heating and cooling light-off curves of methane conversion as a function of temperature on Rhn/Rh1@CeO2 (a) and Rhn/CeO2 (b). Methane conversion curves against the temperature for Rh1@CeO2 and CeO2 are also plotted in (a) for comparison.
A significant discrepancy between Rhn/Rh1@CeO2 and Rhn/CeO2 emerged in their heating light-off curves at temperatures above ~460 °C. Unlike Rhn/Rh1@CeO2, a negative ʹpeakʹ unexpectedly appeared in the 460–800 °C range in the heating light-off curve of Rhn/CeO2, which is abnormal according to the Arrhenius equation24. To shed light on this discrepancy, we collected a Rh K-edge EXAFS spectrum of the used Rhn/CeO2 sample (Fig. S7, Table S1), and found that the Rh NPs have decomposed into single Rh atoms dispersed on CeO2 surfaces following the high-temperature reaction. The results can be further verified by comparing the activity of the used Rhn/CeO2 (the cooling light-off curve in Fig. 2b) with that of Rh1@CeO2 (Fig. 2a). This class of catalyst deactivation by decomposition into single atoms is not unique, and often occurs in supported metal NP-catalyzed reactions, particularly at high temperatures2,21. As expected, the cooling light-off curve of Rhn/Rh1@CeO2 nearly overlaps with the heating curve (Fig. 2a), providing strong evidence for the validity of the single-atom potential confinement strategy in preventing this form of catalyst deactivation. The comparison of activity between 1.5% Rh/CeO2 aged after 800 °C and Rhn/Rh1@CeO2 also confirms the effectiveness of the strategy (Fig. S8), where the Rh loading in both catalysts was similar.
The dynamic evolution of the Rh NPs on Rhn/Rh1@CeO2 and Rhn/CeO2 were directly imaged by using in situ ESTEM in a diluted CH4 + O2 atmosphere in the temperature range of 200–800 °C (see Supplementary materials for more details), as shown in Fig. S9. In Fig. 3, we recorded the ESTEM images of Rhn/Rh1@CeO2 and Rhn/CeO2 at two typical temperatures corresponding to key points on the heating light-off curve of Rhn/CeO2 (Fig. 2b). At 460 °C, the Rh NPs were clearly observed in the ESTEM-SE (secondary electron) images of both samples (insets of Fig. 3a, c), showing the Rh NPs well-dispersed on CeO2, as confirmed by high-resolution ESTEM-ADF (annular dark field) images (Fig. 3a, c). A large difference between the two samples appeared in the ESTEM images taken at the aging temperature of 800 °C. The Rh NPs of Rhn/Rh1@CeO2 can survive after aging at a temperature as high as 800 °C (Figs. 3b, S10). However, the Rh NPs of Rhn/CeO2 have nearly completely disappeared (Fig. 3d), which is consistent with the EXAFS analysis showing isolated Rh atoms after aging (Fig. S7). Hence, this visual evidence further testified to the validity of the single-atom potential confinement strategy under catalytic reaction conditions.

a–d ESTEM-ADF and ESTEM-SE (inset) images of Rhn/Rh1@CeO2 (a, b) and Rhn/CeO2 (c, d) at 460 °C (a, c) or 800 °C (b, d) in a CH4 + O2 atmosphere. Scale bars: 5 nm and 10 nm (insets) in (a, b); 3 nm and 5 nm (insets) in (c, d). One selected typical Rh NP is highlighted by the yellow dashed circle for each image (a–c).
Mechanistic distinction in catalytic activity between Rh nanoparticles and single Rh atoms
Although the single-atom potential confinement strategy has been successful in preventing the dissociation of metal NPs, the use of a highly costly Rh as a precursor inevitably increases the cost for practical applications. Therefore, it is highly desirable to replace precious metals with more affordable alternatives as single-atom sources. However, it is important to clarify that single-atom Rh does not contribute to the low-temperature activity of the Rhn/Rh1@CeO2 system. To address this, we systematically investigated the difference in catalytic activity between the nanoparticles and the single atoms in methane oxidation. First, using DFT calculations combined with phase diagrams, we constructed models of Rh NPs and the single-atom Rh stabilized on CeO2 under realistic reaction conditions (see Note S1 for details, Fig. S11). The density of states (DOS) calculations revealed that Rh NPs exhibit a continuous DOS near the Fermi level (EF) (Fig. S12), suggesting that these Rh clusters have some metallicity. Furthermore, we found that both the valence and conduction bands in this system are mainly contributed by Rh species, which is consistent with the XPS results (Figs. S13, S14). In contrast, the DOS of the Rh1@CeO2 system shows a significant band gap, indicating that the Rh1@CeO2 system remains predominantly non-metallic (Fig. S15). Under these conditions, the valence band is mainly contributed by Rh, while the conduction band has significant contributions from both Rh and Ce species.
Furthermore, we systematically calculated the activation of the first C–H bond in CH4 for both single-atom Rh and Rhn clusters using DFT25,26 (Figs. 4a, S16). The calculated activation barrier for the first C–H bond cleavage on the Rhn/CeO2 is 0.76 eV, which is in good agreement with the experimental Ea value (~0.7 eV, Fig. S6) of Rhn/CeO2 or Rhn/Rh1@CeO2 systems. On Rh1@CeO2, however, a higher activation barrier of 0.91 eV and an exothermic energy release of 0.78 eV were obtained. Moreover, we also calculated the Gibbs free energy profile of methane dissociation at 523 K (Fig. S17), and the results indicate that the methane adsorption becomes significantly more difficult; however, since the entropy effect operates similarly in the methane activation process on the Rh1@CeO2 and Rhn/CeO2 surfaces, the overall activity trend of methane dissociation on different surfaces remains unchanged. Additionally, we analyzed the Bader charges of the active sites (Rh, Ce and O) and CH4 during methane activation over different catalysts (Table S5, Fig. 4b). The calculated results showed that the CH4 molecule transfers 0.23 |e| and 0.24 |e| onto the Rh1@CeO2 and Rhn/CeO2, respectively, from the adsorbed state to the transition state. However, the active Rh atoms only accepts 0.01 |e| in the Rhn/CeO2 system, indicating that the transferred electrons are efficiently redistributed throughout the metal NPs. For Rh1@CeO2, the single-atom Rh barely accepts any electrons from CH4, which are instead accepted mainly by Ce atoms, accompanied by the reduction of Ce4+ to Ce3+ (Fig. 4c, d).

a Calculated energy profiles of the CH4 dissociation on Rhn/CeO2 and Rh1@CeO2; *CH4: original adsorbed state, [CH4]≠: the transition state, and *CH3 + *H: the H3C–H dissociative adsorbed state. b Amount of charge transfer (δQCT) calculated by subtracting the Barder charge at the transition state to that at its original adsorbed state of CH4, the active Rh atom, and the nearest neighboring Ce atom (Fig. S18). The calculated Barder charges are listed in Table S5. c–f Atom-resolution DOS of the Rh, Ce, and O atoms together with CH4 at the original adsorbed state and the transition state ([CH4]≠) on Rh1@CeO2 (c, d) and Rhn/CeO2 (e, f). Blue: Rh; Gray: Ce; Red: O; Black: C; Green: H. All DOSs are aligned with respect to the EF.
To further elucidate the fundamental reasons for the different methane activation properties in the Rh1@CeO2 and Rhn/CeO2 systems, we calculated the DOS at the adsorbed and the transition states for methane activation on these catalysts. For Rh1/CeO2, the lowest unoccupied states are mainly provided by the Rh atoms (Fig. 4c). However, due to the relatively high Rh coordination number (6-coordinated), when methane dissociation occurs at the Rh-O site, the generated H species interact with lattice oxygen, injecting electrons into the system. These electrons remain localized in the empty 4 f orbitals of the neighboring Ce atoms, resulting in the reduction of surface Ce4+ to Ce3+ and the formation of protons. The newly formed Ce3+ exhibits strong repulsive interactions with surrounding species, resulting in a requirement for higher reaction temperatures for C–H bond activation and cleavage (Fig. 4d). By contrast, for the Rh NPs (Fig. 4e), the empty orbital energy levels of the surface Rh and O species are lower than those of the Ce 4 f orbitals, and the coordination number of Rh in this system is three. This suggests that the H species produced by CH4 dissociation first interacts with the surface O− species, injecting an electron such that the empty orbitals of O and Rh gradually disappear, ultimately forming stable OH− species. Hence, the Rh NPs can efficiently accept the electrons transferred from CH4 and then redistribute them to the Rh or O sites within the nanoparticle (Fig. 4f). From the above discussion, we can conclude that CH4 activation on Rh1@CeO2 largely depends on the role of the CeO₂ support, with its catalytic behavior closely resembling the ease of electron acceptance by the Ce 4 f orbitals. The reactivity of this system closely resembles that of CeO2 itself, thus requiring a high temperature to achieve CH4 activation. In contrast, the low-temperature catalytic activity of the Rhn/Rh1@CeO2 system primarily originates from the highly active Rh species present in the Rh NPs, rather than from single-atom Rh, thereby effectively lowering the activation temperature for CH4. Moreover, these results also suggest that the single-atom Rh anchored on the support could potentially be replaced by other appropriate transition metal atoms.
Development of the less costly single-atom potential confinement strategy
It has been reported that, apart from Rh, single transition metal (such as Zr, Mn, Cu, etc.) atoms can be strongly anchored to CeO2 surface Vsites to form stable single-atom catalysts26. Our DFT calculations also suggested that it is energetically favorable to use single Zr atoms as a less costly substitute for precious Rh single atoms to anchor on the CeO2 vacancy sites to form Zr1@CeO2, and the single-atom Zr anchoring can provide potential confinement to prevent the decomposing of metal NPs (Fig. S19).
Using the same method as for Rhn/Rh1@CeO2, we prepared Rhn/Zr1@CeO2 by loading pre-prepared Rh NPs on Zr1@CeO2 (See supplementary Information for details). In Fig. 5a, Rhn/Zr1@CeO2 shows the same activity in the oxidation of methane as Rhn/Rh1@CeO2, indicating that the much less costly Zr atoms play the same role as the precious isolated Rh atoms. An extremely low catalytic activity of Zr1@CeO2 further confirms that Rh NPs are the active sites in Rhn/Zr1@CeO2. We tested the stability of Rhn/Zr1@CeO2 by adopting a canonical protocol2 to approximate realistic conditions, which involved measuring catalytic activity at 460 °C, followed by in situ aging at 800 °C for 1 h, and a subsequent activity measurement at 460 °C (Fig. 5b). Rhn/Zr1@CeO2 showed the same conversion before and after aging, similar to Rhn/Rh1@CeO2 (Fig. S20), whereas Rhn/CeO2 almost completely lost its low-temperature activity following aging. The activity of Rhn/Rh1@CeO2 and Rhn/Zr1@CeO2 is not affected by the water vapor generated from the methane oxidation, even below 500 °C (Figs. S21, S22). Therefore, the use of less expensive transition metals has important implications for the broad application of the single-atom potential confinement strategy.

a Methane conversion as a function of temperature for Rhn/Zr1@CeO2 and Zr1@CeO2 together with Rhn/Rh1@CeO2 for comparison. b Time-on-stream methane conversion profiles for Rhn/Zr1@CeO2 and Rhn/CeO2 with an in situ aging procedure.