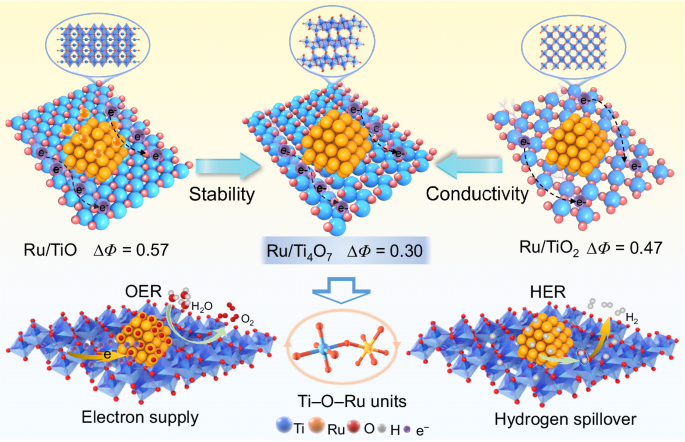
Principles of carrier design
As shown in Fig. 1, non-stoichiometric engineering was employed to finely customize titanium oxide supports (TiO, Ti4O7, and TiO2) with oxygen vacancies of periodic arrangements. Ti4O7, with regularly arranged defects, emerges as a potential competitor with superior structural stability over multi-defect TiO and significantly higher conductivity than TiO2. More importantly, Ti4O7 exhibits a suitable metal-support interaction, which lays the foundation for the dual-function activity of Ru/Ti4O7 in both HER and OER. On the one hand, the higher work function compared to Ru NPs promotes the electron richness of Ru, which can alleviate the dissolution of Ru in OER. On the other hand, compared to TiO and TiO2, the minimum work function difference between Ti4O7 and Ru NPs (∆Φ = 0.30 eV) can reduce the interface Schottky barrier (Supplementary Fig. 1), which promotes electron transport in the composite catalyst and trigger hydrogen spillover during HER.

Schematic diagram of the interaction mechanism between different titanium oxide supports and Ru NPs to activate OER and HER.
Morphology and crystal structure
Ru nanoparticles (NPs) were deposited on titanium oxide supports with different stoichiometric ratios (TiO2, Ti4O7, and TiO) via a wet chemical method (Fig. 2a). Ti4O7 has potential electrocatalytic advantages in support of Ru NPs due to the chemical stability superior to TiO and electrical conductivity far superior to TiO2. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images display the uniform coating of the titanium oxide supports by Ru NPs with a size of ~10 nm (Figs. 2b and 1c). The suitable oxygen vacancies in Ti4O7 promote the Ru3+ adsorption on Ti4O7, which endows the Ru NPs with a more uniform distribution on Ti4O7 compared to TiO2 with low vacancies (Supplementary Figs. 2–6)38,39. In aberration-corrected high-resolution (AC-HRTEM) of Ru/Ti4O7, tight binding and appropriate matching at the nanoscale between the different phases can be observed, accompanied by a smooth transition at the interface (Fig. 2d). The simulated High Angle toroidal dark field image-scanning transmission electron microscope (HAADF-STEM) images obtained through the crystal structure of Ti4O7 and Ru show remarkable agreement with the experimental results (Figs. 2e and 1f). Based on these atomic images, the lattice spacing of 0.377 nm can be attributed to the (1 0 2) plane of Ti4O7 (JCPDS 50-0787), while the lattice spacing of 0.234 nm corresponds to the (1 0 0) plane of Ru (JCPDS 89-4903, Figs. 2g, 1h, and Supplementary Fig. 7). In addition, Ru, Ti, and O elements are uniformly distributed in the energy-dispersive spectroscopy (EDS)-mapping of Ru/Ti4O7 (Fig. 2i), and the characteristic diffraction peaks assigned to Ru and Ti4O7 coexist in the X-ray diffraction (XRD) pattern (Fig. 2j). The above results indicate that Ti4O7 can achieve a suitable interfacial lattice contact with Ru NPs, which provides the possibility for adaptive metal-support interaction and rapid interface electron migration21,40.

a SEM of Ru/Ti4O7. b TEM and c AC-HRTEM images of Ru/Ti4O7. d, e The schematic atom structure and the corresponding simulated HAADF-STEM images of Ti4O7 and Ru in Ru/Ti4O7, respectively. f Crystal plane spacing of Ti4O7 and Ru. g Schematic diagram of interface contact between Ti4O7 and Ru. h EDS mapping of Ru, Ti, and O in Ru/Ti4O7, respectively. i XRD pattern of Ru/Ti4O7.
Electronic and coordination structure analysis
The oxygen vacancy concentration in Ru/TiO2, Ru/Ti4O7, and Ru/TiO shows an increasing trend due to the different stoichiometric ratios of Ti and O (Fig. 3a). The periodically arranged oxygen vacancy not only improves the conductivity of the support but also provides the possibility to form the bridging interfaces between the Ti4O7 and the Ru NPs. In the X-ray photoelectron spectroscopy (XPS) fine spectra of Ru/TiO2, Ru/Ti4O7, and Ru/TiO, the Ti 2p and Ru 3p regions display close overlap (Fig. 3b). The binding energy of Ti 2p in different Ru-loaded titanium oxide materials shows various degrees of shift to the high binding energy (Fig. 3b, and Supplementary Figs. 8–10), which indicates that titanium oxide can serve as an electron donor to promote electron enrichment of Ru (Fig. 3c). As shown in Supplementary Fig. 8c, Ru NPs exhibit only the characteristic peaks of metallic Ru. In the Ru 3d region of Ru/Ti4O7, the signals attributed to Ru–O and Ru–Ru can be observed, corresponding to the interface Ti–O–Ru units41,42. Similar local electronic changes can be further observed in the fine structure of X-ray absorption (XAFS). In the X-ray absorption near-edge structure (XANES), the Ru K-edge also exhibits different white line peak intensity after contacting with various titanium oxide supports (Fig. 3d). Importantly, Ru in Ru/Ti4O7 has the closest valence state to Ru foil compared with that in Ru/TiO2 and Ru/TiO, which is attributed to the low work function difference of Ti4O7 and Ru obtained by the ultraviolet photoemission spectroscopy (UPS) measurements (Supplementary Fig. 1). Meanwhile, Ti pre-K-edge of Ru/Ti4O7 shows a shift to higher energy compared with that of Ti4O7, suggesting that the increased valence state of Ti in Ru/Ti4O7 (Supplementary Fig. 11). The above results indicate that Ru serves as an electron acceptor to form a Schottky contact with the titanium oxide support, and Ti4O7 possesses the best adaptability of the electronic structure with Ru NPs to reduce the interface Schottky barrier. Extended X-ray absorption fine structure (EXAFS) spectra of Ru corresponding k2-weighted EXAFS Fourier transform spectra display a weak Ru–O signal located at 1.92 Å after contacting with TiO2 or Ti4O7 (Fig. 3e). This phenomenon should originate from the interface Ti–O–Ru units between Ru and titanium oxide, which is observed in the wavelet transform more clearly (Fig. 3f). The stable Ti–O–Ru units effectively stabilize the active species and serve as an electron channel to further reduce the interface electron transfer resistance43. As shown in Fig. 3g, the shift of Ti pre-K-edge reflects the changed oxide valence states of Ti in different stoichiometric titanium oxide supports. Furthermore, As illustrated in the k2-weighted EXAFS Fourier transforms of Ti K-edge of Ru/TiO2 and Ru/Ti4O7 (Fig. 3h) and the corresponding wavelet transform (Fig. 3i), the structure belonging to Ti–O–Ru/Ti further verifies the formation of the interface Ti–O–Ru units.

a EPR spectra of Ru/TiO2, Ru/Ti4O7, and Ru/TiO. b XPS fine spectra in Ti 2p and Ru 3p region of Ti4O7, and Ru/Ti4O7. c Schematic illustration of the electronic interaction between Ti4O7 and Ru. d Normalized Ti K-edge XANES of Ti foil, Ru/TiO2, Ru/Ti4O7, and Ru/TiO. e The corresponding k2-weighted Fourier transforms and (f) Wavelet transform of k2-weighted EXAFS signals. g Normalized Ru K-edge XANES of Ru foil, Ru/TiO2, Ru/Ti4O7, and Ru/TiO. h The corresponding k2-weighted Fourier transforms and i Wavelet transform of k2-weighted EXAFS signals.
Electrocatalytic activity and stability evaluation
The electrocatalytic activities of the Ru-loaded titanium oxides were evaluated in a standard three-electrode system to verify the unique advantages of the Ti4O7 support. In the acidic media, Ru/Ti4O7 with optimized Ru loading and annealing temperature exhibits the lowest OER overpotential (150 mV at 10 mA cm−2, Fig. 4a and Supplementary Fig. 12) and Tafel slope (41.26 mv dec−1, Fig. 4b, c). For HER, Ru/Ti4O7 also displays an ultra-low overpotential of 8 mV at 10 mA cm−2 in acidic environments, which is superior to commercial Pt/C and other titanium oxide supported Ru NPs (Fig. 4d). Remarkably, as shown in Fig. 4e, the Tafel slope of Ru/Ti4O7 with minimum work function difference is only 21.24 mV dec−1, which is significantly lower than the value of the conventional Volmer-Heyrovsky/Tafel mechanism (30 mV dec−1)44,45,46. The low work function difference between metal and support is prone to trigger the hydrogen spillover mechanism during HER and provide a low interfacial Schottky barrier and fast interfacial electron migration47. Electrochemical impedance spectroscopy (EIS) further verifies the lowest electron-transfer resistance of Ru/Ti4O7 (Supplementary Fig. 13). Notably, the superior difunctional activity of Ru/Ti4O7 also exhibited scalability in the neutral and basic environments (Fig. 4c, f, and Supplementary Figs. 14–17). In addition, Ru/Ti4O7 exhibits the highest double-layer capacitance (Cdl) value (68.95 mF cm−2) with the same Ru loading, suggesting that more exposed Ru active sites of the Ru/Ti4O7 compared to Ru/TiO2 and Ru/TiO (Supplementary Figs. 18 and 19). The LSV polarization curves normalized by Cdl and the mass of Ru indicate that the active Ru sites in Ru/Ti4O7 have the highest specific activities (Supplementary Fig. 20) and mass activity (Supplementary Fig. 21). The outstanding bifunctional performance of Ru/Ti4O7 exceeds that of most of the excellent bifunctional catalysts in the recent reports (Supplementary Tables 2, 3), especially in acidic environments (Fig. 4g).

a Linear sweep voltammetry (LSV) polarization curves of OER and b Tafel plots of Ru, commercial RuO2, Ru/TiO2, Ru/Ti4O7, and Ru/TiO (loading amount: 1 mg cm−2) in 0.5 M H2SO4 (pH = 0.3). c The corresponding OER overpotential at 10 mA cm−2 in different pH environments. d LSV polarization curves of HER and (e) Tafel plots of Ru, commercial Pt/C, Ru/TiO2, Ru/Ti4O7, and Ru/TiO (loading amount: 1 mg cm−2) in 0.5 M H2SO4 (pH = 0.3). f The corresponding HER overpotential at −10 mA cm−2 in different pH environments. g Comparison of overpotential with that of other bifunctional catalysts for water splitting in 0.5 M H2SO4 recently reported. h Chronopotentiometry curves of Ru/Ti4O7 at 10 mA cm−2, and −10 mA cm−2 in 0.5 M H2SO4.
The stability of Ru/Ti4O7 was validated via chronopotentiometry. In severe acidic environments, Ru/Ti4O7 demonstrates surprising stability of OER and HER for 500 h with a negligible activity decay (Fig. 4h) and slight structural collapse (Supplementary Fig. 22), which exceeds the operating life of Ru/TiO (Supplementary Fig. 23). Ru/Ti4O7 also requires lower potentials to drive OER compared to Ru/TiO2, contributing to the lower energy consumption of the water electrolysis devices over a long period (Supplementary Fig. 23). More importantly, Ru/Ti4O7 can maintain stable operation for 300 h at a high current density of 200 mA cm−2 with only slight activity decay after 3000 cyclic voltammetry (CV) cycles (Supplementary Figs. 24 and 25). The lower current response in the CV corresponding to the oxidation-reduction of Ru in Ru/Ti4O7 compared to Ru and Ru/TiO implies the inhibited oxidation of Ru by Ti4O7 (Supplementary Fig. 26). Supplementary Fig. 27 manifests that the Ru and Ti loss of Ru/Ti4O7 gradually slows down during long-term OER. Ti gradually dissolves in the first 50 h of cycling, which is attributed to the reaction between the unstable surface of the electrode. After 50 h, the dissolution rate of Ti slows down due to the stabilization of the surface of the electrode. Furthermore, the characteristic diffraction peaks belonging to Ru and Ti4O7 can still be observed in the XRD pattern of Ru/Ti4O7 after OER without new characteristic peaks compared to the apparent dissolution of Ru/TiO (Supplementary Fig. 28). As shown in the electron paramagnetic resonance (EPR) spectra (Supplementary Fig. 29) and XPS spectra of O 1 s before and after stability tests (Supplementary Fig. 30a), a slight decrease in oxygen vacancies in the Ti4O7 support is observed, indicating the structural stability of the Ti4O7 support under high current densities. In addition, the almost unchanged XPS signals of Ti and Ru with slight shift towards high binding energy after OER further provide evidence for the good structural maintenance of Ru/Ti4O7 during the reaction (Supplementary Fig. 30b). The excellent stability of Ru/Ti4O7 is attributed to inhibited oxidation process, which should be derived from the electron enrichment of Ru through the stabilized Ti–O–Ru units.
Origin of enhanced difunctional activity of Ru/Ti4O7
A series of in-situ tests were performed in acid environments to trace the origin of the activity and stability of Ru/Ti4O7. In-situ XAFS explored the structural evolution of Ru/Ti4O7 during OER. Ru/Ti4O7 underwent 20 CV cycles to obtain the stable surface before recording the in-situ XAFS spectra. In the most severe acidic environments, the intensity of the white line of the Ru K-edge increases with the applied potentials of OER (Fig. 5a), indicating the oxidation of Ru NPs. Furthermore, in the Fourier transform R-space (Fig. 5b), the gradually increased signal intensity of the Ru–O path verifies the formation of RuOx. Notably, the peaks attributed to metallic Ru–Ru can always be observed even under the high potentials of OER, which corresponds to the Ru metallic phase shown in the XRD pattern of Ru/Ti4O7 after OER. The above results indicate that the RuOx species locally formed on the Ru/Ti4O7 can act as a barrier layer to slow down the further oxidation and dissolution of Ru NPs. The in-situ XAFS spectra show that the Ti K-edge of Ti4O7 almost completely overlaps under the OER potentials without prominent oxidation characteristics, which benefits from the stable structure of Ti4O7 (Supplementary Fig. 31a). The R-space of Ti further verifies that the Ti–O bond in Ti4O7 does not change significantly during OER (Supplementary Fig. 31b). The stability of Ru/Ti4O7 can be more verified in the explicit valence states of Ti, and Ru. The valence state corrected by the normalized white line intensity of the corresponding reference of Ti in Ru/Ti4O7 changes slightly between 3.52 and 3.61 with the application potentials growing (Supplementary Fig. 32). Meanwhile, the average valence state of Ru increases from 0.98 to 1.64. The significant change does not appear until the potential reaches 1.7 V vs. RHE (Fig. 5c). This phenomenon further verifies that the Ru NPs still maintain structural stability under high potentials of OER. The incomplete oxidation of Ru NPs is beneficial to maintaining the outstanding conductivity of the composite material during the electrocatalytic process. In the HRTEM image of Ru/Ti4O7 after OER, the tight interfacial contact between Ru NPs and Ti4O7 can still be maintained (Supplementary Fig. 33). In addition, a locally amorphous structure can be observed in the outer layer of Ru NPs in Ru/Ti4O7 after OER, which should correspond to the RuOx species (Fig. 5d).

a Normalized in-situ Ru K-edge XANES of Ru/Ti4O7. b The corresponding k2-weighted Fourier transforms. c The valence states of Ru and Ti in Ru/Ti4O7 obtained via Ru K-edge under different potentials. d HRTEM of Ru/Ti4O7 after OER. In-situ ATR-SEIRAS spectra of (e) Ru and f Ru/Ti4O7 for OER in 0.5 M H2SO4 under different potentials vs. RHE. g Intensity difference of the infrared signals at 1038 and 1189 cm–1. h Current responses to pulse voltammetry for Ru and Ru/Ti4O7. i Relationship between charge storage and potential of Ru and Ru/Ti4O7. j Schematic illustration of the optimized OER process induced by the Ti4O7 support.
Moreover, the reaction mechanism of OER was revealed by in-situ attenuated total reflection-surface enhanced infrared absorption spectra (ATR-SEIRAS). Figure 5e shows that, as the bias increases to 1.4 V vs. RHE, in-situ ATR-SEIRAS spectra of Ru exhibit absorption bands around the vibration frequency of 1038 cm−1, corresponding to the *OOH/*OO intermediates. Simultaneously, a distinct absorption signal at the vibration frequency of 1189 cm−1 gradually strengthens with the application of OER potential, corresponding to *O-O* in the LOM. In-situ ATR-SEIRAS spectra of Ru/Ti4O7 appear at similar positions as Ru, attributing to the absorption bands of *OOH/*OO (Fig. 5f). The broadening and shifting of absorption peaks may originate from the transition of the deprotonation process. Furthermore, almost no signal corresponding to *O-O* is observed around the vibration frequency of 1190 cm−1. Additionally, the normalized density difference of in-situ ATR-SEIRAS spectra corresponding to *OOH/*OO and *O-O* signals is presented in Fig. 5g to determine the proportion occupied by adsorption evolution mechanism and LOM in the acidic OER process38. The higher adsorption density difference implies a higher proportion of adsorption evolution mechanism in the reaction process. In the high potential range, Ru undergoes surface reconstruction to form RuOx species, enabling both the adsorption evolution mechanism and LOM to drive OER. Meanwhile, in-situ ATR-SEIRAS spectra of Ru/Ti4O7 consistently exhibit a higher adsorption density difference than Ru during OER. This phenomenon indicates that Ru/Ti4O7 tends to drive OER through the adsorption evolution mechanism, which can realize more stable OER compared with the LOM with potential catalyst dissolution. In the pH-dependence tests, compared with Ru NPs, Ru/Ti4O7 showed less correlation with acidity, which verified that it was more inclined to drive OER with adsorption evolution mechanism (Supplementary Fig. 34). These results demonstrate that Ru/Ti4O7 tends to drive OER through the surface adsorption evolution mechanism, which can realize more stable OER compared with the LOM with potential catalyst dissolution38,48.
Pulse voltammetry test was employed to assess the deprotonation capability of catalysts, which can confirm the source of the enhanced activity of Ru/Ti4O7 in OER49,50. Under different voltage pulses (Supplementary Fig. 35), Ru and Ru/Ti4O7 exhibit alternating cathodic and anodic current pulses (Fig. 5h). The oxidation charge storage capacity of different catalysts was further measured by integrating the anodic current response to voltage pulses. As shown in Fig. 5i, Ru/Ti4O7 demonstrates a higher oxidation charge storage capacity compared to Ru NPs, implying that Ru/Ti4O7 undergoes a faster deprotonation process to form reaction intermediate *O49. These findings suggest that the appropriate metal-support interactions between Ti4O7 and Ru can activate Ru sites by promoting the deprotonation process in OER. Furthermore, in the EIS bode plots, the pre-OER process of Ru/Ti4O7 results in an uneven distribution of surface charges (Supplementary Fig. 36), which is manifested by a reduction in the frequency peaks within the range of 1.40-1.45 V vs. RHE and a shift towards higher frequencies compared to the broader transition phase peaks of Ru NPs (1.35–1.45 V vs. RHE). This phenomenon suggests that Ru/Ti4O7 exhibits a faster charge dissipation to accelerate deprotonation during OER to reaction kinetics51. In summary, the moderate metal-support interactions between Ru NPs and Ti4O7 through Ti–O–Ru units can reduce interface Schottky barriers to accelerate electron transfer and achieve electron enrichment at Ru sites to slow down the corrosion of Ru during OER52,53,54. In addition, electronic modulation of active Ru sites facilitates the deprotonation process of OER (Fig. 5j). During the HER process, EIS Nyquist plots at different current densities were recorded and fitted by the equivalent circuit inset of Supplementary Fig. 37a. R2 reflects the hydrogen adsorption resistance on the material surface in the equivalent circuit13,44. Hydrogen adsorption kinetics were quantified by plotting logR2 versus overpotential and calculating the EIS-derived Tafel slopes. As shown in Supplementary Fig. 37, the reduced slope of Ru/Ti4O7 of 21.3 mv dec−1 compared with that of Ru/TiO2 and Ru/TiO indicates the accelerated hydrogen adsorption kinetics, which is related to the hydrogen spillover (Supplementary Fig. 38, and Supplementary Table 4). The above results further demonstrate that Ru/Ti4O7 achieves fast HER kinetics through a potential hydrogen spillover mechanism.
Theoretical calculation for intrinsic activity and stability analysis
Density functional theory (DFT) calculations reveal the impact of the support adaptability of Ti4O7 on the electronic structure modulation and the bifunctional reactivity of the Ru sites. The energy band structure manifests that Ti4O7 and TiO have a rich density of state distribution at the Fermi level, symbolizing their superior conductivity compared to a noticeable band gap of TiO2 (Supplementary Fig. 39). The differential charge density shows a significant yellow area near the Ru atoms at the interface between Ru and Ti4O7, which verifies the electron enrichment of Ru atoms (Supplementary Figs. 40 and 36c)55. In the average potential profile (Supplementary Fig. 41d), the electrostatic potential difference between Ru and Ti4O7 can be more explicitly observed. Subsequently, Pourbaix diagrams were utilized to verify the stabilization of Ru based on Ti–O–Ru units between Ru and titanium oxide supports. According to the Pourbaix diagram of Ru–O (Fig. 6a), Ru is prone to over-oxidation to form high-valent Ru4+ at high potentials for OER, especially in acidic environments. Furthermore, the robust interface Ti–O–Ru units effectively increases the theoretical dissolution potential of Ru in electrochemical oxidation as shown in the Pourbaix system of Ti–O–Ru (Fig. 6b). In Fig. 6c, the Bade and valence state calculations at the interface show that the Ru sites at the Ru/Ti4O7 interface have the highest valence state (−0.007), which is closest to the initial valence state of Ru and consistent with the XAFS results. This electronic structure adaptability gives Ru/Ti4O7 a lower interface Schottky barrier compared to Ru/TiO2 and Ru/TiO to accelerate interface electron migration and trigger the hydrogen spillover mechanism for HER47.

a Ru–O, and (b) Ti–O–Ru Pourbaix diagrams generated with aqueous ions concentration of 10−4 M at 25 °C. c Bade charge at the interface of Ru/TiO2, Ru/Ti4O7, and Ru/TiO and corresponding calculated valence states of Ru and Ti. d Free energy profiles of different OER intermediates at 0 V and 1.23 V for Ru site in Ru, Ru/TiO2, Ru/Ti4O7, and Ru/TiO. e Free energy profiles of HER intermediates for Ru site in Ru, Ru/TiO2, Ru/Ti4O7, and Ru/TiO.
The structural model corresponding to the partially oxidized Ru NPs loaded on Ti4O7 was constructed to calculate the OER pathways based on the adsorption evolution mechanism (Supplementary Figs. 41–43). The local oxidation combined with the electron redistribution between Ru and different titanium oxide supports endow the Ru sites with varying intrinsic activities through the d-band center modulation. The downward shift of the d-band center reduces the energy of the antibonding orbitals formed by the adsorption reaction intermediates and the d-orbitals of Ru, which implies the weak adsorption of reaction intermediates48,56. Therefore, in the surface oxidized Ru/TiO, Ru/TiO2 and Ru/Ti4O7, the d-band centers of the Ru sites are −1.80 eV, −1.81 eV and −2.07 eV, respectively (Supplementary Fig. 44 and Supplementary Table 5), which endow the corresponding *OH adsorption free energies with −0.482 eV, 0.435 eV, and 0.703 eV at U = 0 V, respectively (Fig. 6d). Subsequently, the deprotonation process is also optimized. Ru/Ti4O7 exhibits the smallest deprotonation free energy and the lowest theoretical potential for OER (ΔG = 1.540 eV, Supplementary Table 6). The enhanced deprotonation process is consistent with experimental results. The catalysts drive HER in its initial structure at the reduction potentials (Supplementary Fig. 45)21,57. The free energy step diagram verifies that the weak HER activity of Ru sites in Ru NPs originated from the strong adsorption of HER intermediates (Fig. 6e). Specifically, the d-band center of Ru at the interface exhibits distinct variations with the interface electron migration induced by the contact between Ru and different titanium oxides. The d-band center of Ru is −1.72 eV. After contact with TiO, TiO2, and Ti4O7, the corresponding d-band centers of Ru shift to −1.77 eV, −1.89 eV, and −1.98 eV, respectively (Supplementary Fig. 46). Owing to the downward shift compared to the original Ru NPs induced by the metal-support interaction, the Ru sites in Ru, Ru/TiO, Ru/TiO2, and Ru/Ti4O7 have *H adsorption free energies of −0.745 eV, −0.446 eV, −0.171 eV, and 0.123 eV, respectively (Fig. 6e, Supplementary Table 7)58,59. Therefore, Ru/Ti4O7 has the lowest theoretical reaction potential of 0.123 eV for HER. The theoretical calculations further verified that the appropriate support can promote the intrinsic activity of the Ru sites for both OER and HER.
Performance of the water electrolysis device
Firstly, the unique advantages of suitable support design were verified through membrane-free water electrolysis tests. As shown in Fig. 7a, the water electrolysis driven by Ru/Ti4O7 with excellent bifunctional activity only requires a cell voltage of only 1.44 V to reach a current density of 10 mA cm−2 with stable operation for 300 h (Fig. 7b) in the acidic environments. Noteworthily, the corresponding Faraday efficiency is also close to 100%. This excellent water splitting performance exceeds that of the state-of-the-art difunctional catalysts recently reported in the acidic media (Fig. 7c). The superiority of the Ti4O7 support for water splitting in the pH-universal environments has also been verified (Supplementary Figs. 47, 48 and Supplementary Table 8). Benefitting from the advantages of conductivity and stability, Ru/Ti4O7 was used as both cathode and anode catalysts in the MEA electrolyzers (Fig. 7d). For the PEM water electrolysis (Fig. 7e), Ru/Ti4O7 ‖ Ru/Ti4O7 exhibits significantly lower cell voltage than that of RuO2 ‖ Pt/C and maintains long-term operation of 200 h at 200 mA cm−2 and 300 h at 500 mA cm−2 (Fig. 7f and Supplementary Fig. 49), which is superior to most recently reported Ru-based catalysts (Supplementary Table 9). The unique structural advantages of the PEM electrolyzer and the characteristics of high-temperature operation combined with the excellent stability of Ru/Ti4O7 achieve robust PEM water electrolysis60. The EIS Nyquist curves of the MEA further verified the conductivity advantage of Ru/Ti4O7, which significantly reduces the energy consumption of the electrolyzer under high current densities (Supplementary Fig. 50)61. Furthermore, Ru/Ti4O7 also possesses extensible high activity and stability in AEM (Supplementary Fig. 51). The apparent advantages of bifunctional Ru/Ti4O7 in membrane electrode assembly show impressive industrialized application prospects.

a LSV polarization curves of Ru ‖ Ru, RuO2 ‖ Pt/C, Ru/TiO2 ‖ Ru/TiO2, Ru/Ti4O7 ‖ Ru/Ti4O7, and Ru/TiO ‖ Ru/TiO (loading amount: 1 mg cm−2) in 0.5 M H2SO4 (pH = 0.3) for the acidic water splitting. b Stability tests of Ru/Ti4O7 ‖ Ru/Ti4O7 at 10 mA cm−2, and the inset display the corresponding Faraday efficiency at 100 mA cm−2. c Comparison of the cell voltage and stability of Ru/Ti4O7 ‖ Ru/Ti4O7 with those of recently reported bifunctional catalysts for acidic water splitting. d Conceptual model of the PEM water electrolyzer flow cell using pure water as feedstock. e Steady polarization curves of Ru/Ti4O7 ‖ Ru/Ti4O7 and RuO2 ‖ Pt/C (loading amount: 2 mg cm−2) for PEM water electrolyzer in pure water (pH = 6.7) at 80 °C. f Stability tests of Ru/Ti4O7 ‖ Ru/Ti4O7 for PEM water electrolyzer at 200 mA cm−2.