Catalyst design and reaction optimization
For the past few decades, organometallic complexes based on phosphine ligands have revolutionized organic synthesis47,48,49,50,51,52,53,54,55,56,57,58,59,60. A number of phosphine-based complexes have been designed and applied for various synthetic processes, including coupling, hydrogenation, hydroformylation, carbonylation, amination, and C–H activation reactions1,2,3,4,5,6,47,48,49,50,51,52,53,54,55,56,57,58,59,60. This achievement is largely attributed to the use of specific multidentate phosphines, which have been well recognized as valuable ligand frameworks47,48,49,50,51,52,53,54,55,56,57,58,59,60. Particularly, bi- and tri-dentate phosphine ligated complexes have emerged as promising hydrogenation and amination catalysts47,48,49,50,51,52,53,54,55,56,57,58,59,60. For instance, we and others reported the applications of such complexes for hydrogenative cross-coupling of nitriles to amines57, reductive hydrolysis of nitriles to alcohols58, hydrogenation of CO259, carboxylic acids or esters60 and amides7,8,9,10,11. Inspired by these works, we started to design a suitable metal-phosphine complex as a potential catalyst for the hydrogenation of primary amides to prepare primary amines. To explore an appropriate amide hydrogenation catalyst to work under mild conditions, initially, we tested in situ-generated different metal-phosphine complexes for the reaction of benzamide 1 in the presence of 10 bar molecular hydrogen at 115 °C (Fig. 3 and Supplementary Tables 1 and 2). Initially, we tested xantphos (L1), tripodal triphos (L2) and tetraphos (L3) as representative commercially available di-, tri-, and tetra-dentate ligands. Except for cobalt, all the tested phosphine complexes based on non-noble metals were completely inactive (Fig. 3 and Supplementary Table 1, entries 1-21). Co-L2 showed little activity with the formation of an undesired benzyl alcohol 3 (Fig. 3 and Supplementary Table 1, entry 11). On the other hand, Ru-phosphine complexes, particularly Ru-tripodal triphos (L2), showed good activity (Fig. 3 and Supplementary Table 1, entry 23) and resulted in a conversion of 75% with the formation of 10% of the desired product 2 and remaining side-products such as benzyl alcohol 3 (30%) and N-benzylidenebenzylamine 4 (35%). However, the selectivity of the Ru-L2 system could not be improved to obtain satisfactory yields of 2. Among Pd, Pt, and Rh complexes, Pd-based complexes are completely inactive, while Pt and Rh complexes yielded 7–24% of cyclohexanecarboxamide as an undesired product (Supplementary Table 1, entries 25–33).

Reaction conditions: 0.5 mmol benzamide, 4 mol% metal salt, 5 mol% ligand, 10 bar H2, 3 mL TFE, 115 °C, 24 h, conversion and yields were determined by GC analysis using mesitylene as standard. *Same reaction conditions in the presence of 5 bar NH3. Fe-salt = Fe(BF4)2 ·6H2O, Mn-salt = Mn(OTf)2, Co-salt = Co(BF4)2·6H2O, Ni-salt = Ni(OTf)2, Cu-salt = Cu(BF4)2·6H2O, Ru-salt = Ru(acac)3, Pd-salt = Pd(OAc)2, Pt-salt = PtCl2, Rh-salt = RhCl3.
At this point, we considered it worthwhile to prepare substituted tripodal triphos ligands and test their corresponding Ru-complexes in the model reaction. Accordingly, p-methoxy and p-methyl substituted triphos ligands (Triphos(p-anisole); L4 and Triphos(p-tolyl); L5) were prepared, and the resulting Ru-L4 and Ru-L5 in situ complexes were tested for their activities and selectivities. Interestingly, both ligands (L4 and L5) showed excellent activities and achieved complete conversion of 1 with the formation of the desired product 2 up to 55% (Fig. 3 and Supplementary Table 2, entries 2a–3a). However, in both cases, lower selectivity toward 2 was achieved, with the formation of other side-products such as benzyl alcohol 3 (12–15%) and N-benzylidenebenzylamine 4 (32–34%). Remarkably, the use of ammonia suppressed the formation of side-products 3 and 4, and up to 98% of the desired product benzylamine 2 was obtained using Ru-L4 system (Fig. 3 and Supplementary Table 2, entry 5b). Next, to know the performance of other phosphine ligands, commercially available mono-, bi-, and tri-dentate ligands (L6–L10) were tested. All these ligands generated non- or less-active Ru-complexes (Supplementary Table 3). Further different Ru-precursors were tested with L4 and observed that Ru(acac)3 is the suitable one to generate the most active and selective catalyst (Supplementary Table 4). All these results confirmed that Ru(acac)3–L4 is the best catalyst system to convert benzamide to benzylamine in quantitative yield in the presence of molecular hydrogen and ammonia. Subsequently, all catalytic reactions were carried out with Ru-L4 in the presence of H2 and NH3. The model reaction was performed in different solvents using Ru-L4, and the hydrogenation of 1 proceeded well in trifluoroethanol (TFE) and hexafluoroisopropanol (HFIP) to obtain a maximum yield of 2 (Supplementary Table 5). The fluorinated solvent TFE is more acidic and exhibits strong hydrogen bonding activity due to the electron-withdrawing effect of the fluorine atoms, which facilitates the solubility of hydrogen as well as the activation of the substrate, amide or imine. In other tested solvents we observed less conversion of the starting material (6–12%) (Supplementary Table 5).
To further optimize the reaction conditions, important parameters such as the amount of catalyst, ligand, reaction temperature, pressure of hydrogen and reaction time were evaluated (Supplementary Tables 6–9). The best results were obtained with 4 mol% of catalyst (1:1.2 ratio of Ru(acac)3 and L4), in the presence of 10 bar hydrogen and 5 bar NH3 at 115 °C in 24 h (Supplementary Tables 6–9).
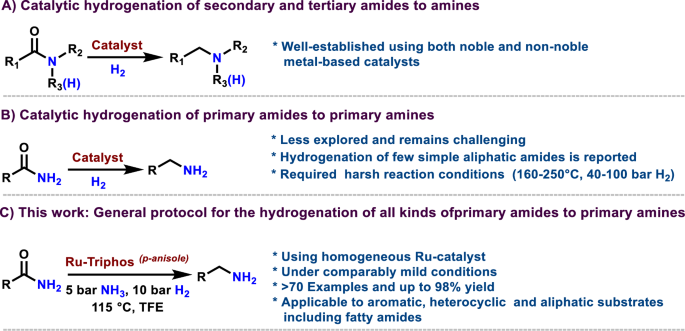
Hydrogenation of aromatic primary amides
After successful design of the optimal catalyst, Ru(acac)3-Triphos(p-anisole) (Ru-L4) and reaction conditions for the model reaction, we subsequently demonstrated the broad applicability of this Ru-based protocol for the hydrogenation of all kinds of primary amides to synthesize benzylic, heterocyclic, and aliphatic primary amines as well as fatty amines (Figs. 4–7). At first, simple, substituted and functionalized aromatic amides were hydrogenated and obtained corresponding benzylic amines in up to 96% yield (Fig. 4). Benzamides substituted with methyl-, t-butyl, and -phenyl groups as well as naphthamide were reduced to corresponding benzylic amines (Fig. 4, products 5–12). Interestingly, the hydrogenation of terephthalamide, an important diamide, was smoothly proceeded to give p-xylylenediamine (Fig. 4, product 13). Diamines are used in a variety of industrial applications, including the production of polymers and advanced materials. Next, amine-, N, N-dimethyl, hydroxyl-, ether-, trifluoro-, sulfonate-, and boronic acid ester-substituted benzamides were reduced to provide corresponding functionalized benzylamines in yields up to 93% (Fig. 4, products 14–27). Halogen-substituted benzylamines are highly valuable and are increasingly used as important precursors and intermediates in organic synthesis. Preparation of such compounds from corresponding amides under hydrogenative conditions, without significant dehalogenation, is obviously important. Gratifyingly, Ru-L4 complex was able to hydrogenate fluoro-, chloro-, and bromo-substituted benzamides to corresponding halogenated amines in up to 91% yield (Fig. 4, products 28–35). However, the iodo-group was not tolerated under experimental conditions. Likewise, other functional groups such as nitro, nitrile, keto and C-C double bonds were not tolerated, and all these functionalities were reduced in addition to the amide group (Supplementary Fig. 8). In the case of methyl-ester substituted benzamide, the transesterification was observed with trifluoroethanol solvent along with the reduction amide group.
![Fig. 4: Ru-catalyzed hydrogenation of aromatic amides to benzylic amines[a].](https://media.springernature.com/lw685/springer-static/image/art%3A10.1038%2Fs41467-026-69794-2/MediaObjects/41467_2026_69794_Fig4_HTML.png?as=webp)
Reaction conditions: [a]0.5 mmol amide, 4 mol% Ru(acac)3, 5 mol % L4, 10 bar H2, 5 bar NH3, 3 mL TFE, 115 °C, 24 h, isolated yields. [b] Same as [a] at 125 °C. Products were isolated as free amines and converted to hydrochloride salts. Corresponding hydrochloride salts were subjected to NMR analysis.
![Fig. 5: Hydrogenation of heterocyclic carboxamides using Ru-L4 catalytic system[a].](https://media.springernature.com/lw685/springer-static/image/art%3A10.1038%2Fs41467-026-69794-2/MediaObjects/41467_2026_69794_Fig5_HTML.png?as=webp)
Reaction conditions: [a]0.5 mmol amide, 4 mol% Ru(acac)3, 5 mol% L4, 10 bar H2, 5 bar NH3, 3 mL TFE, 115 °C, 24 h, isolated yields. [b] Same as [a] at 125 °C. Products were isolated as free amines and converted to hydrochloride salts. Corresponding hydrochloride salts were subjected to NMR analysis.
![Fig. 6: Ru-L4 catalyzed hydrogenation of aliphatic amides[a].](https://media.springernature.com/lw685/springer-static/image/art%3A10.1038%2Fs41467-026-69794-2/MediaObjects/41467_2026_69794_Fig6_HTML.png?as=webp)
Reaction conditions: [a]0.5 mmol amide, 4 mol% Ru(acac)3, 5 mol% L4, 10 bar H2, 5 bar NH3, 3 mL TFE, 115 °C, 24 h, isolated yields. [b] Same as [a] at 125 °C. cGC yield using mesitylene as a standard. Products were isolated as free amines and converted to hydrochloride salts. Corresponding hydrochloride salts were subjected to NMR analysis.

Reaction conditions: 0.5 mmol amide, 4 mol% Ru(acac)3, 5 mol % L4, 10 bar H2, 5 bar NH3, 3 mL TFE, 115 °C, 24 h, isolated yields. Products were isolated as free amines and converted to hydrochloride salts. Corresponding hydrochloride salts were subjected to NMR analysis.
Hydrogenation of heteroaromatic primary amides
The synthesis of heterocyclic amines is of considerable importance because these products have widespread applications in organic synthesis and drug discovery as well as bio-molecular chemistry. Different hetero-aromatic primary amides were subjected to hydrogenation in the presence of Ru-L4, and corresponding heterocyclic primary amines were produced in up to 91% yield (Fig. 5). Nicotinamide is a medicinally relevant compound, as well as isonicotinamide and picolinamide were successfully hydrogenated to provide corresponding picolylamines in up to 89% yields (Fig. 5, products 36–42). Picolylamines serve as potential ligands in organometallic complexes as well as precursors for making functional materials and organic molecules. In addition, amine-and bromo-substituted picolylamines were also obtained from corresponding amides (Products 39–42).
Apart from pyridine carboxamides, quinoline carboxamide, pyrazine carboxamide and indole carboxamide, as well as furan carboxamide, reacted well and gave corresponding heterocyclic amines in up to 93% yields (Products 43–49). Other interesting amines such as (2-(piperidin-1-yl)phenyl)methanamine (50) and (4-((4-ethylpiperazin-1-yl)methyl)phenyl)methanamine (51) were also prepared from corresponding primary amides.
Hydrogenation of aliphatic primary amides
Inspired by the results with (hetero)aromatic substrates, we turned our attention to the hydrogenation of aliphatic amides. As shown in Fig. 6, a series of phenyl-aliphatic as well as aliphatic primary amides were successfully reduced using Ru-L4 and produced respective primary amines in up to 91% yield.
Notably, this Ru-based amide reduction methodology led to the preparation of biogenic amines, which play an essential role in the physiological function of the human body as well as in the nervous system and blood pressure control61,62,63. As an example, phenethylamine (PEA), a trace amine that acts as a central nervous system stimulant, was prepared in 89% yield (product 52). Likewise, Tyramine (catecholamine releasing agent), Dopamine (neuromodulatory molecule), and β-Methylphenethylamine (stimulant and human TAAR1 agonists) were synthesized in 85–86% yields (Products 53-55). Additionally, bromo-, chloro-, and phenyl- substituted phenethylamines were obtained in excellent yields (products 56-58). Histamine, a biological compound that is involved in local immune responses, communication and neurotransmission as well as regulating physiological functions of the human body63, was also prepared from the corresponding primary amide (Product 61). Aliphatic amides such as cyclohexanecarboxamide, adamantane-1-carboxamide, and tetrahydro-2H-pyran-4-carboxamide, as well as pentane and heptane amides, reacted well, providing respective aliphatic amines in up to 91% yields (Products 63–67). Further, this Ru-catalyzed hydrogenation methodology is also amenable to the synthesis of hexamethylenediamine, a key feedstock for nylon 66 polymer, directly from adipamide (product 68).
Synthesis of fatty amines
Fatty amines are an important class of oilfield chemicals that find increasing applications in industrial and daily life products64,65,66,67,68,69. For example, they are widely used as asphalt additives, anticaking agents, surfactants, fabric softeners and other household materials64,65,66,67,68,69. Given their importance, the production capacity of fatty amines is >1 million tons per year, and their market volume is expected to grow from USD 3.44 billion in 2021 to USD 4.87 billion in 2026, at a compound annual growth rate of 7.1%67,68. Commonly, fatty amines are produced by the hydrogenation of nitriles64,69. Alternatively, we demonstrate here that these amines can be prepared from fatty amides, which in turn are produced starting from vegetable oils and fats. As a result, applying Ru-L4, 7 fatty primary amines were synthesized in up to 78% yield (Fig. 7).
Activity and selectivity of Ru-triphos(p-anisole) for the hydrogenation of secondary and tertiary amides
To investigate the performance of Ru-L4 in the hydrogenation of secondary and tertiary amides (Supplementary Fig. 9), the reduction of three selected substrates, N-(4-methoxyphenyl)benzamide (88; secondary amide), N-methylbenzamide (93; secondary amide) and 4-methoxy-N,N-diphenylbenzamide (95; tertiary amide), was carried out under similar experimental conditions that were used for the hydrogenation of primary amides (4 mol% Ru(acac)3, 5 mol% L4; 10 bar H2, 115 °C, TFE, 24 h). All these substrates underwent hydrogenation, and 50–96% conversion was obtained. However, significantly lower selectivities (40–52%) were obtained to produce the corresponding secondary and tertiary amines. In addition, the formation of side products such as the corresponding imines, alcohols and C-N bond cleavage products was observed. Notably, when the reaction of 86 was performed in the presence of ammonia, a high yield (87%) of primary amine 2 was achieved (Supplementary Fig 8., reaction 4).
Proposed reaction pathway and mechanism for the Ru- triphos(p-anisole) catalyzed hydrogenation of primary amides
After having demonstrated the synthetic applications, we became interested in knowing the reaction pathway(s) and mechanism of this Ru-L4 catalyzed amide hydrogenation protocol. Following the reaction progress of the model system at different intervals of time revealed the in situ formation of benzyl alcohol 3 and N-benzylidenebenzylamine 4 in addition to the desired benzylamine 2 (Supplementary Table 9). The generated benzyl alcohol 3 and N-benzylidenebenzylamine 4 were completely converted into 2 within 24 h, and an overall yield of 98% of the desired product 2 was achieved. To investigate the reaction behavior of alcohol, benzyl alcohol 3 was reacted with ammonia in the presence of Ru-L4 at 115 °C in TFE for 24 h (Supplementary Fig. 10, reaction 1). This reaction resulted in 62% conversion of 3 and afforded 21% benzylamine 2 and 40% N-benzylidenebenzylamine 4. Interestingly, the reaction of N-benzylidenebenzylamine 4 in the presence of hydrogen and ammonia with Ru-L4 produced 98% yield of benzylamine 2 (Supplementary Fig. 10, reaction 2). However, in the absence of ammonia, N-benzylidenebenzylamine 4 yielded 92% of the hydrogenated product, dibenzylamine 85% and 5% benzylamine 2 (Supplementary Fig. 10, reaction 3). As expected, benzamide 1 showed no conversion in the absence of molecular hydrogen (Supplementary Fig. 10, reaction 4).

Path A=C–O bond cleavage pathway. Path B=C–N bond cleavage pathway.
All these results suggest that the corresponding alcohol 3 and the secondary imine 4 are formed as stable intermediates in this Ru-catalyzed primary amide hydrogenation process. Based on these observations, a general reaction pathway is proposed and shown in Fig. 8. First, the primary amide (A) is reduced to hemiaminal (B) in the presence of Ru-L4 and molecular hydrogen. Subsequently, B undergoes either C–O bond (path A) or C–N bond (path B) cleavage reaction. In pathway A, dehydration of B takes place via C-O bond cleavage in the presence of Ru-L4 to generate primary imine (C), which is then reduced to the desired primary amine (D). In addition, C can react with D to form the corresponding secondary imine (G), which could be hydrogenated to the secondary amine (Fig. 2 (H)). However, in the presence of ammonia, it is further converted to the primary amine (D). In path B, hemiaminal (B) underwent hydrogenative deamination via C–N bond cleavage to form alcohol (F) in the presence of hydrogen. Afterwards, F reacts with ammonia in the presence of Ru-L4 to form benzylamine (D). In this amide hydrogenation, ammonia plays significant roles: (a) it shifts the equilibrium between intermediates C (primary imine) and G (secondary imine) toward C to favor the formation of D (benzylamine), (b) it allows the amination of the alcohol, and (c) helps to transform the secondary imine (G) to the primary imine (C) and finally toward D. Thus, the use of external ammonia is crucial for this Ru-catalyzed amide hydrogenation process to produce primary amines with high yields and selectivities.
NMR spectroscopic studies
To elucidate the formation of the ruthenium complex and the corresponding dihydride complex, we performed in situ variable temperature high-pressure NMR (VT-HP NMR) experiments. Since the hydrogenation of primary amides to primary amines with Ru-L4 proceeded well in both TFE and HFIP as solvents with similar conversions and yields, and deuterated HFIP was commercially available, it was chosen as the solvent for the NMR spectroscopic investigations. For convenience, the commercially available Triphos L2 ligand was used instead of the synthesized ligand L4.
Firstly, a solution of [Ru(acac)3] (16.7 mM) and Triphos (L2) (16.7 mM) in a dry and degassed 1:1 mixture of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) and 1,1,1,3,3,3-hexafluoro-2-propanol-d2 (HFIP-d2) was transferred to a sapphire high-pressure NMR cell under argon and then pressurized with 5 bar NH3 and 10 bar H2. Since the reaction could not be monitored for a long period of time in the spectrometer, the remaining solution was transferred to a 10 mL Swagelok reactor, where the reaction proceeded for 24 h. The reaction output was then evaluated by ex situ NMR. The 1:1 mixture of deuterated and non-deuterated HFIP was chosen to allow both the lock of the magnetic field and the detection of possible hydride species. The in situ NMR experiment showed that the activation of the metal precatalyst resulted rather slowly, and after 2 h at 115 °C, only a minor fraction of the expected [Ru(Triphos)]-motif was detectable together with traces of hydride species (Supplementary information; Section S4). As we suspected a possible coordination of NH3, the reaction mixture from the reactor after 24 h was not purged with argon. Instead, the pressure was slowly released until 1.4 bar, and the obtained pale-yellow solution was transferred to a Young-NMR tube under Ar. Under these conditions, a portion of dissolved gases usually still remains in solution. Traces of a very thin and dark precipitate were observed in the NMR tube, suggesting some metal deposition. The ex situ 1H- and 31P-NMR spectra showed the formation of a main complex almost quantitatively. Its resonance at δ = 29.9 ppm in the 31P-NMR spectrum was accompanied only by a small portion of non-coordinated ligand with a resonance at δ = −27.3 ppm (Supplementary Fig. 14). The 1H spectrum showed a highly intense signal for dissolved NH3 at δ = 3.59 ppm. No signals at all were detected in the hydride region. Attempts to isolate this complex failed, since the complex readily decomposed just during the concentration of the solution in vacuum. In order to characterize this complex in solution, the experiment in the reactor was repeated with slightly modified conditions: [Ru(acac)3] (11.7 mM) and Triphos (12.6 mM) were dissolved in HFIP-d2, since no hydride signals were detected at the end of the reaction, and the mixture was heated under same pressure and temperature as before for 24 h.
The 31P-NMR spectrum showed the expected complex almost quantitatively formed (Fig. 9a). A very small portion of non-coordinated ligand (~4%) was detected. A 1H–31P HMBC allowed to confirm the signals of the -CH3 and the P-bound -CH2– groups at δ = 1.75 and 2.69 ppm, respectively, arising from the coordinated Triphos ligand. Also, the cross peaks showing the coupling of the aromatic protons were visible. Interestingly, a small coupling with a proton resonance at δ = 3.1 ppm was detected. A 1H–15N HMBC confirmed that this signal corresponds to coordinatively bound NH3 (Fig. 9b). Because of the proximity and the intensity of the resonance arising from dissolved ammonia, it was not possible to get an accurate integral value for the signal of coordinated NH3 at δ = 3.1 ppm. Since the resonances arising from hydroxy and amino groups show a dynamic behavior, the NMR sample was transferred back to the reactor and purged 4 times with 20 bar argon. The again measured solution did not show any changes in the 31P-NMR spectrum, but to our delight the signal corresponding to coordinated NH3 shifted from 3.1 to 2.74 ppm, while the less intense, but still strong signal corresponding to dissolved ammonia shifted from 3.72 to 4.72 ppm (Supplementary Fig. 15). A new 1H–15N HMBC confirmed the shift of the coordinatively bound NH3 (Supplementary Fig. 16) and the integral value showed clearly the presence of 2 × NH3, which corresponds to a [Ru(Triphos)(NH3)2] complex.

a 1H and 31P{1H} quantitative NMR spectra after the reaction of Ru(acac)3 with Triphos in reactor under NH3/H2. The stars indicate the signals for the CH3 and the P-CH2 groups of coordinated Triphos. The dot indicates coordinating NH3. Reaction conditions: [Ru] = 11.7 mM, [Triphos] = 12.6 mmol, solvent: HFIP-d2, p(NH3) = 5 bar, p(H2) = 10 bar, ϑ = 115 °C, reaction time = 24 h. b 1H-31P and 1H-15N HMBC NMR spectra after the reaction of Ru(acac)3 with Triphos in reactor under NH3/H2. The stars indicate the signals for the CH3 and the P-CH2 groups of coordinated Triphos. The dot indicates coordinating NH3. Reaction conditions: [Ru] = 11.7 mM, [Triphos] = 12.6 mmol, solvent: HFIP-d2, p(NH3) = 5 bar, p(H2) = 10 bar, ϑ = 115 °C, reaction time = 24 h.
In situ and ex situ NMR spectroscopic investigations of the ruthenium-catalyzed hydrogenation of benzamide confirmed that the same [Ru(Triphos)(NH3)2] complex is formed in the presence of substrate (Supplementary information; Section S4.4). The NMR measurements during the hydrogenation of benzamide showed an almost identical behavior concerning the formation of the molecular complexes as during the preformation of Ru(acac)3 under H2/NH3. Our experiments before shown that the use of ammonia suppressed the formation of side products and enhanced the selectivity towards the formation of the primary amine (Fig. 3). These results together with the NMR studies show that a critical concentration of NH3 is necessary for the reaction to occur selectively, under the complex [Ru(Triphos)(NH3)2] II exist as a resting state of the catalyst, which can be transformed by H2 into the active hydride species. The oxidative addition of molecular hydrogen to [Ru(Triphos)(NH3)2] with simultaneous dissociation of an ammonia ligand would yield the ruthenium(II) dihydride complex I, which, upon dissociation of the second ammonia ligand, would produce the unsaturated 16-electron complex III as the active catalytic species70 (Fig. 10).

Showcasing the generation of Ru-ammonium complex, Ru-dihydride as active catalytic species and reaction pathways via C–O and C–N bond cleavage.
These results led us to propose the catalytic cycle for this Ru-L4-catalyzed hydrogenation of primary amides to primary amines, which is shown in Fig. 10. The ruthenium dihydride complex III reacts with the primary amide A to give the coordinated hemiaminal complex IV. At this point, C-O or C-N bond cleavage took place to provide the primary imine C or alcohol F, respectively. In a separate cycle, F reacts with ammonia in the presence of III and generates C. As the final step in the catalytic cycle, the primary imine C is hydrogenated by complex III to furnish the desired primary amine D with the regeneration of III.