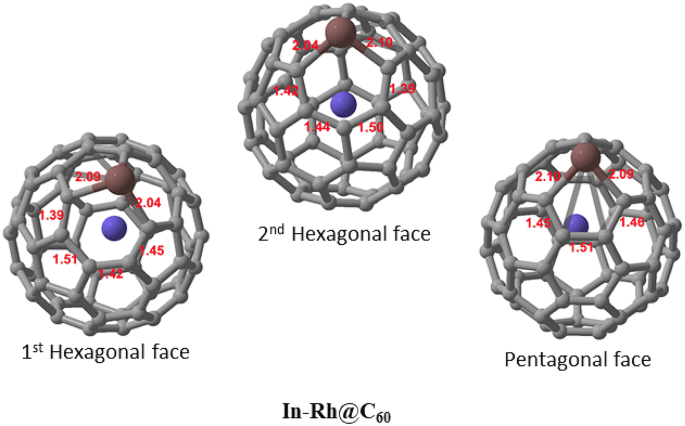
Structural geometry
The optimized molecular structure of In-Rh@C60 is depicted in Fig. 1. Fullerene C60 is a three-dimensional (3D) carbon structure that consists of 60 covalently bonded carbon atoms arranged in a spherical shape. It is made up of 12 pentagonal faces and 20 hexagonal faces which allow C60 to form a stable, cage-like structure. Herein, the C60 nanocage was structurally doped with In, while Rh was encapsulated within its hollow cage structure. These structural modifications influence the system’s electronic properties and its nature of interactions with gas molecules, such as NH3, NO, and NO2 upon adsorption. The C-In bond lengths in the optimized structure are 2.09 Å, 2.04 Å, and 2.10 Å, with an average of 2.08 Å. The pentagon face of the system exhibits C-C bond lengths of 1.45 Å, 1.51 Å, and 1.46 Å, resulting in an average of 1.47 Å. Meanwhile, the average C-C bond lengths in both the first and second hexagonal faces adjacent to the In atom are 1.44 Å.

Optimized structures of the modified In-Rh@C60 system showing the first and second hexagonal faces, as well as the pentagonal face.

Optimized structures of the gas-adsorbed In-Rh@C60 systems upon interaction with NH3 and NO (with two adsorption sites considered), showing the first and second hexagonal faces, as well as the pentagonal face.
As shown in Table 1, upon adsorption of NH3, the average C-In bond length in the NH3-In-Rh@C60 system measured 2.09Å, indicating no significant deviation from the modified system (In-Rh@C60). Similarly, the average C-C bond lengths of the first and second hexagonal faces were measured at 1.45 Å and 1.44 Å, respectively, while that of the pentagonal face was placed at 1.47 Å, respectively (see Fig. 2). This suggests that the interaction of NH3 with the system does not affect the electronic structural properties of the modified system. After the adsorption of NO at the N-site, the average C-In bond length in the NO-N-In-Rh@C60 system measured 2.14 Å, with an increase of 0.06 Å over that of the modified system. This indicates that a strong interaction exists between NO and In at the N-site of adsorption on the system. This can be attributed to the polarity of NO. The average C-C bond lengths of both the first and second hexagonal faces adjacent to the In atom gave a negligible difference from the original system. Similarly, that of the pentagonal face resulted in a 0.01 Å reduction from the modified system. This suggests that despite the strong interaction between NO and In, it had no significant effect on the overall carbon network of the system.
The adsorption of NO at the O-site resulted in a 0.03 Å increase in the C-In bond length, indicating an influence on the NO-O-In-Rh@C60 system. In atom has a low electronegativity while the nitrogen atom in NO gas is electronegative. The resulting polarization of the C-In bond could rationalize the changes in the bond length. Whereas, both the hexagonal and pentagonal faces resulted in no difference, indicating that the polarization does not affect the ordered carbon network.
Furthermore, when NO2 was adsorbed at the N-site, the average C-In bond length resulted in a remarkable 0.11 Å increase from the original system, indicating a significant influence on the NO2-N-In-Rh@C60 system (see Fig. 3). NO2 has a bent molecular geometry due to the presence of a lone pair of electrons on the nitrogen atom which repels the oxygen atoms, compared to the linear molecular geometry of NO. The molecular structure and properties of NO2 may be responsible for the stronger interactions with In and a more significant change in the C-In bond length, compared to NO. Moreover, since it has a higher electronegativity, the resulting polarization between the nitrogen atom of NO2 and In may be responsible for the stronger electrostatic interactions with the site of adsorption on the system. Again, the average C-C bond lengths of both hexagonal and pentagonal faces resulted in no changes. This suggests that despite the strong interaction observed between NO2 and In, the carbon network of the system remained unaffected.
Additionally, upon the interaction of NO2 at the O-site, a 0.09 Å increase in the C-In bond length was observed, indicating a significant impact on the NO2-O-In-Rh@C60 system. Similarly, the high polarity between the electronegative nitrogen atom in NO2 and the relatively electropositive atom can be attributed to the strong interaction observed in the C-In bond length of the system. Meanwhile, the average C-C bond length of both the hexagonal and pentagon faces showed no significant changes in the carbon network despite the strong electrostatic interaction observed upon the adsorption of NO2 onto the system. Therefore, the increasing order of changes observed in the C-In bonds is as follows: NH3-In-Rh@C60 < NO-N-In-Rh@C60 < NO-O-In-Rh@C60 < NO2-N-In-Rh@C60 < NO2-O-In-Rh@C60. Overall, we observe that chemical properties, such as electronegativity and polarity, molecular structure, and atomic size play a vital role in the geometry of the In-Rh@C60 system upon interactions with these nitrogen-containing gases.

Optimized structures of the gas-adsorbed In-Rh@C60 systems upon interaction with NO2 at N and O sites, showing the first and second hexagonal faces, as well as the pentagonal face.
Coordination and spin States stability
Evaluating the spin state and coordination environment is essential for determining the electronic structure, stability, and reactivity of metal-doped systems48. For the studied system (In-Rh@C60), these factors determine its adsorption and catalytic properties. Different spin states represent the different electronic configurations, which affect the total energy of the system49. The lowest spin energy state indicates the most stable electronic configuration under given conditions and can predict the optimal spin state for adsorption studies50. The coordination of In and Rh on C60 influences the stability and adsorption of the gas molecules. By examining the different spin states – singlet, triplet, quintet and septet, the most stable electronic configuration can be determined, thus, influencing the modified system’s reactivity towards gas adsorption. The spin states energies were calculated in electronvolts (eV) and presented in Table S2. The most stable spin state was the triplet state with an energy of 0 eV, while quintet state has a spin state energy of 0.682 eV, indicating a high stable configuration. The septet and singlet states have the highest spin state energy of 2.557 eV and 1.120 eV, respectively, indicating that transition to these states leads to a less stable configuration. The energetic feasibility of the triplet state suggests that adsorption studies should focus primarily on the spin configurations to capture the most realistic electronic properties of In-Rh@C60. However, upon adsorption of NO and NO2 gases, the spin polarization effect may consequently shift the system towards a higher spin state (quintet) as a result of unpaired electrons in these gas molecules. These findings show that adsorption and catalytic activity on the modified In-Rh@C60 system should be primarily analyzed in the triplet and quintet states, since they dominate the electronic structure and reactivity of the system. If the adsorbed system is stable at higher spin state like quintet, desorption may occur due to the system’s tendency to return back to the triplet stable or ground state. Thus, stronger adsorption, alongside high charge transfer and spin transition, can lead to a spontaneous desorption process.
Reactivity and stability analysis
The calculated values of the lowest unoccupied molecular orbital (LUMO), highest occupied molecular orbital (HOMO), and energy gap for the modified In-Rh@C60 system and its derivatives after adsorption are presented in Table 2. The study of frontier molecular orbital (FMO) offers valuable insights into the electronic properties, chemical reactivity, and interactions of systems with other molecules. This quantum mechanics technique revolves around the highest occupied molecular orbital and the lowest unoccupied molecular orbital. The energy gap which is the energy difference between the HOMO and LUMO is vital in understanding the properties of material, such as conductivity, electrical, thermal, reactivity, and optical properties51,52. Here, the energy gap study, which is the energy difference between the HOMO and LUMO of the system and its derivatives provided an understanding of the crucial properties, such as conductivity, reactivity, and stability. The HOMO and LUMO plots upon the adsorption of gases are depicted in the Fig. 4. The quantum descriptors presented in Tables 2 and S3 were computed using the Koopman’s approximation given by Eqs. (2–8).
$$\:\text{I}\text{P}={-E}_{\text{H}\text{O}\text{M}\text{O}}$$
(2)
$$\:EA={-E}_{\text{L}\text{U}\text{M}\text{O}}$$
(3)
$$\:\text{E}\:\left(\text{e}\text{V}\right)\hspace{0.17em}=\hspace{0.17em}\text{E}\:\left(\text{H}\text{a}\right)\:\times\:\:27.2114$$
(4)
$$\:\mu\:=-\raisebox{1ex}{$1$}\!\left/\:\!\raisebox{-1ex}{$2$}\right.\left({E}_{\text{H}\text{O}\text{M}\text{O}}+{E}_{\text{L}\text{U}\text{M}\text{O}}\right)$$
(5)
$$\:H=\raisebox{1ex}{$1$}\!\left/\:\!\raisebox{-1ex}{$2$}\right.\left(IP-EA\right)=\frac{{E}_{\text{L}\text{U}\text{M}\text{O}}-{\text{E}}_{\text{H}\text{O}\text{M}\text{O}}}{2}$$
(6)
$$\:S=\frac{1}{2{\upeta\:}}=\frac{1}{IP-EA}=\frac{1}{{\text{E}}_{\text{L}\text{U}\text{M}\text{O}}-{\text{E}}_{\text{H}\text{O}\text{M}\text{O}}}$$
(7)
The electrophilicity index (\(\:\omega\)), was calculated using the equation given as:
$$\:\omega\:=\frac{{\mu\:}^{2}}{2\text{H}}$$
(8)
The calculated electronic properties of the In-Rh@C60 system and its derivatives, determined using two levels of theory, PW6B95-D3/GenECP (Method 1) and ωB97X-D/LANL2DZ (Method 2), reveal distinct trends in HOMO, LUMO, and energy gap values, providing insights into their stability, reactivity, and potential for gas sensing applications. The results from both methods indicate consistent trends for specific systems while highlighting differences in their approximation of electronic properties, which merit detailed analysis.
From the results obtained using Method 1, the energy gap values for the systems ranged from 0.705 eV for NO-O-In-Rh@C60 to 1.537 eV for NO2-O-In-Rh@C60. The notably small energy gap of NO-O-In-Rh@C60 suggests higher reactivity due to its lower stability and increased ease of electron excitation. This behavior may be attributed to the destabilizing interaction between NO and the In-Rh@C60 framework at the O-site of adsorption. Conversely, NO2-O-In-Rh@C60, with the largest energy gap, exhibits greater stability and reduced electronic reactivity, possibly due to charge transfer interactions and electronic structural modifications induced by NO2 at the O-site53,54. The stability trends observed from Method 1 imply that NO2-O-In-Rh@C60 is the most stable system, whereas NO-O-In-Rh@C60 and NO-N-In-Rh@C60, with their narrower energy gap values, are the least stable and most reactive.
In comparison, Method 2 yielded energy gap values that are significantly larger across all systems, ranging from 3.152 eV for NO-O-In-Rh@C60 to 5.166 eV for NO2-N-In-Rh@C60. This increase in energy gap values highlights the tendency of Method 2 to overestimate electronic stability of the systems, predicting larger energy gap values compared to Method 1. Interestingly, while the trends for NO-O-In-Rh@C60 and NO2-O-In-Rh@C60 remained consistent with Method 1, Method 2 reversed the ranking of NO2-N-In-Rh@C60, suggesting it to be the most stable system. This discrepancy highlights the influence of the theoretical approach on the interpretation of electronic properties, with Method 1 potentially offering a more realistic approximation for reactive systems, given its incorporation of dispersion corrections.
The trends observed for HOMO and LUMO energies further substantiate these conclusions. For both methods, NO-O-In-Rh@C60 consistently exhibited the least negative HOMO value, indicating its high reactivity and low ionization potential. On the other hand, NO2-O-In-Rh@C60 showed the most negative LUMO energies, consistent with its high electron affinity and stability. These properties reflect the interplay between the electronic structure of In-Rh@C60 and the adsorbed gases, which modulate conductivity, stability, and reactivity based on the nature of the interaction and the adsorption site. The implications of these findings for gas sensing applications are significant. NO2-O-In-Rh@C60, with its high stability and moderate conductivity, emerges as the optimal candidate for long-term sensing applications where durability is critical. Conversely, NO-O-In-Rh@C60, with its superior conductivity, is better suited for dynamic sensing environments, albeit with limitations in long-term stability. The close energy gap values for NH3-In-Rh@C60 and NO2-O-In-Rh@C60, as predicted by Method 1, suggest comparable electrical and chemical properties, further supporting their potential utility in stable sensing applications. Generally, the comparison of the two methods highlights the advantages of PW6B95-D3/GENECP (Method 1) in providing a balanced approximation of stability and reactivity, particularly for systems with pronounced electronic interactions. Method 2, while emphasizing stability, may underestimate reactivity and conductivity for certain systems. These differences emphasize the importance of selecting appropriate theoretical methods for studying electronic properties, especially for systems like fullerene. The observed variations in the energy gap values across both methods reinforce the role of adsorption-induced electronic modifications in determining the suitability of these systems for gas sensing applications, offering a pathway for further experimental validation and optimization.

HOMO and LUMO plots of the gas-adsorbed systems.
Density of States (DOS)
The Density of States (DOS) serves as a crucial descriptor in evaluating the electronic properties and reactivity of molecular systems. It provides insights into the contributions of atomic orbitals to the electronic structure, particularly in adsorption and charge transfer processes55. In this study, the DOS calculations were performed to further elucidate the electronic interactions within the modified fullerene and its functionalized systems upon interaction with NH3, NO, and NO2 gases. This analysis was conducted using the two DFT methods of this study (PW6B95-D3/GEnECP and ωB97X-D/LANL2DZ) to compare the accuracy of these methods in describing the stability and reactivity of the studied systems. Figures 5 and 6 present the DOS plots, computed using the Multiwfn 3.7 package and visualized with Origin software. For Method 1 (PW6B95-D3/GEnECP), the DOS plots of the systems show a dominant carbon atom peak at the Valence Band Maximum (VBM), reflecting a high electronic contribution. At the Conduction Band Minimum (CBM), carbon and Rh peaks were more pronounced, indicating their strong charge acceptance abilities, with minor contributions from In. Strong In-Rh hybridization at the VBM suggested that the system exhibited good adsorption properties, as Rh near the Fermi level could participate in charge transfer. This observation is consistent with studies on transition metal-functionalized silicon carbide nanotubes (TM@SiCNTs), where similar charge transfer interactions were reported for cobalt, Rh, and iridium in trichloromethane gas adsorption56. It is worth noting that carbon exhibited the highest peak at the VBM and CBM indicating its ability to donate and accept charges, thus contributing to the stability of the system. Upon interacting with NH3 molecule, a shift in the peaks was observed, as an overlap (hybridization) of the Rh, In, N and O peaks was observed at the VBM with a slight elevation of N peak, indicating its tendency to donate charges and promote electron localization. At the CBM, Rh exhibited the highest peak, followed by In revealing their tendency to accept charges. During the interaction of the modified In-Rh@C60 system with NO2 at both sites of adsorption (N and O), the O atom peak was observed to be slightly elevated in the VBM region, indicating a potential for charge transfer, while an overlap was observed between N, Rh and In atoms. This behavior agrees with previous findings on metal-doped fullerenes, where gallium and In dopants introduced new electronic states near the Fermi level, enhancing charge transfer and adsorption57. In the CBM region, Rh and In peaks were more pronounced, while an overlap between N and O peaks was observed, indicating high reactivity and catalytic activity. Upon interaction with NO gas at N and O, peaks were observed to overlap each other and slightly elevated in the VBM region, depicting a measure of charge transfer. An overlap between Rh, In, N, O was observed near the Fermi level. However, in the CBM region, Rh and In were observed to possess the highest peak, with N & O peaks overlapping each other. This indicates stronger conductivity especially for sensing and adsorbing NO gas.
For Method 2 (ωB97X-D/LANL2DZ), the carbon peak was equally observed to have the highest peaks at both VBM and CBM. The DOS plots of the systems reveal hybridization between Rh and In peaks in the VBM region, while Rh and In peaks were elevated in the CBM region, indicating high charge acceptance. Upon adsorption of NH3, all the peaks of each fragment overlapped, with a slight elevation of N & O peaks in the VBM region. Meanwhile, Rh, In, and H peaks were elevated in the CBM region. Upon adsorption of NO2 gas at the two sites of adsorption, the O atom peak was observed to be slightly elevated, while hybridization was observed between Rh, In, and N peaks in the VBM region. On the other hand, in the CBM region, In and Rh had high peaks while N and O overlapped. Upon adsorption of NO gas at both sites of adsorption, all the atom fragments overlapped in the VBM region, accompanied by a slight elevation of N and O peaks near the fermi level. In the CBM region, Rh and In exhibited the highest peaks, while an overlap between N and O was observed.

Density of states plots of the modified In-Rh@C60 system and gas-adsorbed In-Rh@C60 systems calculated at the DFT/PW6B95-D3/GENECP level of theory (Method 1).

Density of states plots of the modified In-Rh@C60 system and the corresponding gas-adsorbed systems calculated at the DFT/ωB97X-D/LANL2DZ level of theory (Method 2).
Electron localization function (ELF)
The Electron Localization Function (ELF) analysis provides crucial insights into electron density distribution and bonding characteristics of the adsorbed gases with In-Rh@C59 system (Figs. 7 and 8). According to Akman et al.., 2023, ELF values less than 0.5 signify delocalization of electrons, while ELF values greater than 0.5 indicate localization of electrons58. The ELF scale for the DFT/PW6B95-D3/GEnECP and DFT/ωB97X-D/LANL2DZ methods ranges from 0 (blue regions, representing delocalized electrons) to 1 (red regions, representing highly localized electrons or strong covalent bonding), while the intermediate values indicate polar or shared interactions. The ELF for both methods shows similar charge localization trends, confirming a consistent charge transfer mechanism. Generally, from the ELF-mapped plot of the first DFT method, the Rh atom exhibits moderate electron localization (0.5 – 0.8) suggesting a partially covalent bond with the C59 system. The In atom, with a blue color, signifies delocalization, which implies the transfer of charges from the doped atom to the fullerene system. The carbon-to-carbon interactions of the fullerene framework display strong electron localization (0.8 – 1.0), which reflects its sp2-hybridized center. Some regions of the fullerene cage show lower localization (0.4 – 0.6), which signifies modified π-electron delocalization due to Rh and In doping. The presence of the adsorbed molecules alters the ELF-mapped plot. ELF near the adsorbed molecule ranges from 0.4 to 0.9, signifying partial covalent bonding and electron transfer. This charge redistribution enhances the adsorption activity of the systems. Partial electron localization near the system’s adsorption sites confirms active charge transfer, while the presence of strong covalent bonding within the fullerene and weaker interactions at the adsorption site (dopants) ensures stability and reactivity.

Electron Localization Function (ELF) analysis of the modified In-Rh@C₅₉ system and the corresponding gas-adsorbed systems calculated at the DFT/PW6B95-D3/GENECP level of theory. The ELF plots illustrate electron density distribution, highlighting bond localization and charge delocalization at the adsorption sites.

Electron localization function (ELF) analysis of the modified In-Rh@C59 system and the corresponding gas-adsorbed systems calculated at the DFT/ωB97X-D/LANL2DZ level of theory. The ELF plots illustrate electron density distribution, highlighting bond localization and charge delocalization at the adsorption sites.
Adsorption of gases on the modified In-Rh@C60 fullerene system
Previously, the adsorption of NO and NO2 gas molecules has been studied theoretically to determine the intricate properties of the bare C60 fullerene. In line with this, many reports have been made regarding the adsorbing potency of the bare C60 fullerene, as well as their functionizedsystems59,60,61. In this study, the C60 fullerene was structurally modified by encapsulating the Rh atom within its hollow cage-like structure and doping it through the substitution of one carbon atom with an In atom. The newly designed Rh-encapsulated In-doped C60 fullerene was then used in the adsorption of NH3, NO, and NO2 by considering two sites of adsorption in the case of NO and NO2. The most stable adsorption configurations were identified, and the results obtained using Eq. (1) are presented in Tables 3 and 4. In all cases, chemisorption was obtained for the labeled systems, showing that the designed system is a promising adsorbent material for NH3, NO2, and NO2 gas molecules. In the following sections, we examine the adsorption of these gases individually to evaluate the influence of sites on the adsorption.
NH3 adsorption on the modified In-Rh@C60 system
Considering the N-site only, NH3 gas was adsorbed on the modified In-Rh@C60 system, with a high adsorption energy of -12.11 eV. This energy obtained depicts a chemisorption phenomenon and indicates that the NH3 gas molecules were strongly adsorbed on the modified system of In-Rh@C60. The relatively greater energy shows that among the studied adsorbates, NH3 will be strongly adsorbed on the modified system compared to the adsorption of the NO gas molecules.
NO adsorption on the modified In-Rh@C60 system
Due to the presence of N and O atoms of the NO gas molecules, the N- and O-sites of adsorption were considered during the adsorption process. The adsorption energies of -8.397 and -7.690 eV corresponding to the N- and O-sites of adsorption were obtained upon adsorption. Both values indicate strong adsorption of NO gas on the In-Rh@C60 system. Since the adsorption at the N-site shows greater energy than at the O-site, the N-site exhibited relatively stronger adsorption.
NO2 adsorption on the modified In-Rh@C60 system
In the adsorption of NO2 on the modified In-Rh@C60 system, the adsorption energy falls within a close range of -13.49 eV to -12.15 eV, where the highest energy is attributed to the adsorption at the O-site. Since a more negative adsorption energy indicates stronger adsorption62, the adsorption configuration at the O-site exhibits the most favorable adsorption compared to the N-site of adsorption. In all cases, the strongest adsorption was seen in NO2-O-In-Rh@C60. Generally, the adsorption follows this trend: NO-N-In-Rh@C60 < NO-O-In-Rh@C60 < NO2-N-In-Rh@C60 < NO2-O-In-Rh@C60, representing an increasing order of adsorption strength.
Compared to Method 2 (DFT/ωB97XD/LANL2DZ), the adsorption energies are higher, which signifies strong interactions between the gases and the modified system. However, the limitation is the desorption process might be difficult due to the strong bonds between the adsorbate and the adsorbent. Thus, the adsorption energy of Method 2 is preferable based on the reactivity studies, as it indicates higher chemical reactivity and a more favorable adsorption-desorption process.
Thermodynamic analysis
The thermodynamics properties of the studied systems were calculated at the DFT/ωB97XD/LANL2DZ level of theory to further elucidate their adsorption properties, as well as the strength and stability of interactions between the gases (NH3, NO, and NO2) and the modified system (In-Rh@C60). The calculations for all systems were performed at an absolute temperature of 298.15 K (250 C) and standard pressure of 1 atm, as presented in Table S4. The spontaneity and feasibility of the interactions between In-Rh@C60 and NH3, NO2, and NO gases can be determined using thermodynamic parameters, thereby confirming the potential of the modified system for detecting and sensing of the pollutant gases63. Thermodynamics parameters such as enthalpy change (ΔH0) and Gibbs free energy change (ΔG0) are key indicators for determining the thermal stability and spontaneity of the interactions between In-Rh@C60 and NH3, NO2, and NO gases64. The electronic energy (Ɛ0), zero-point energy correction (Ɛ_ZPE), thermal correction to energy (Etot), thermal correction to enthalpy (Hcorr), thermal correction to free energy (Gcorr), Ɛ0 + zero-point Energy, Ɛ0 + thermal Energy correction, Ɛ0 + thermal enthalpy correction, Ɛ0 + thermal free energy correction, change in Gibbs free energy (ΔG0), and change in enthalpy (ΔH0) are all presented in Table S4. The change in Gibbs free energy (ΔG0) and change in enthalpy (ΔH0) associated with the adsorption of the gas molecules onto the modified system were calculated using the following mathematical expression:
$$\:{\varDelta\:H}_{ads}={H}_{system}-({H}_{gas}+{H}_{surface})$$
(3)
$$\:{\varDelta\:G}_{ads}={G}_{system}-({G}_{gas}+{G}_{surface})$$
(4)
Where Hsystem is the enthalpy of the gas-adsorbed system, Hgas is the enthalpy of the individual gas molecules (NH3, NO2, and NO), Hsurface is the enthalpy of the modified system, Gsystem is the Gibbs free energy of the gas-adsorbed system, Ggas is the Gibbs free energy of the the individual gas molecules (NH3, NO2, and NO), and Gsurface is the Gibbs free energy of the modified system. The electronic energy (Ɛ0) shows that NO2-O-In-Rh@C60 is the most stable, with the highest value of -69746.1 eV. In contrast, In-Rh@C60 has the least Ɛ0 value, indicating an increase in stability upon interaction with the gas molecules. NH3-In-Rh@C60 exhibits the highest Etot and Hcorr, suggesting higher thermal energy involvement, which may influence its practical stability. NO-O-In-Rh@C60 shows the least Gcorr value of 8.9852 eV, indicating a thermodynamic preference for adsorption-based applications. The calculated ΔG0 reveals that NH3-In-Rh@C60 exhibits the highest positive value, indicating an endothermic process, non-spontaneous and thermodynamically unfavorable adsorption process. In contrast, NO-N-In-Rh@C60 and NO-O-In-Rh@C60, with ΔG0 values of -0.0716 and -0.1208 eV, respectively, indicate a spontaneous adsorption process. Similarly, ΔH0 analysis reveals that NH3-In-Rh@C60 and NO2-O-In-Rh@C60 undergo an endothermic adsorption process with enthalpy values of 2.2176 and 0.0936 eV, respectively. However, NO-N-In-Rh@C60 and NO-O-In-Rh@C60 exhibit an exothermic process, with enthalpy values of -0.0523 and -0.0046 eV, respectively, which implies the spontaneity of adsorption process. These results suggest that In-Rh@C60 has a greater tendency to detect and sense NO gas molecules under standard temperature and standard pressure (298.15 K, 1 atm).
Visual studies
Non-covalent interaction studies
Non-Covalent Interaction (NCI) studies as applied to this investigation between adsorbates and adsorbents give relevant insights into the nature of weak intermolecular forces governing adsorption phenomena65. They offer a broad basis for understanding non-covalent interactions such as van der Waals forces, hydrogen bonding, π-π stacking, and electrostatic interactions, which play crucial roles in adsorbate-adsorbent interactions66. In this context, we utilized DFT to analyze the electron density and energy landscapes associated with adsorption processes. By visualizing the spatial distribution of non-covalent interactions, we identified regions of attractive and repulsive forces between adsorbates and adsorbents, thereby providing vital insights into the binding geometries and adsorption energetics67. It has been reported that this analysis allows for the quantification of interaction energies, enabling the prediction of adsorption strengths and selectivity for different adsorbate-adsorbent pairs68. Based on our NCI results depicted in Fig. 9, all the studied systems displayed large green patches in the mesoporous of the fullerene C60 material. This could be influenced by the encapsulation of Rh, a transition metal exhibiting nucleophilic behavior. Thus, the green color depicts the van der Waals force of interactions, thereby contributing to the system’s stability behavior. Whereas the red color captured in the intramolecular bonding of the systems depicts the steric force of repulsion. The two forces enhance the system’s capability to adsorb gases from the environment. Conversely, NO2-O-In-Rh@C60 (adsorbent binding to the O-site of NO2 gas) was observed with steric and strong attraction (H-bonding) in its intermolecular regions, implying greater adsorption compared to NO2-N-In-Rh@C60 (adsorbent binding to the N-site of NO2 gas) which did not display any electrostatic force in its intermolecular bonding. Examining the adsorption of NO gas, at both the N- and O-sites, the intermolecular regions were notable with blue patches, which depicted strong attraction or H-bonding. This indicates strong interaction at any site of the NO gas molecules, offering strong hydrogen bonding between the studied modified system and the adsorbate (gas). A similar character was also observed when the studied system interacted with NH3. Hence, the insights gleaned from this study can play a crucial role in designing and optimizing materials for sensor applications, given the investigation of diverse electrostatic forces. This highlights the significance of this work in advancing sensor technology.
QTAIM analysis
The Quantum Theory of Atoms in Molecules (QTAIM) is well known for investigating interactions between adsorbates and adsorbents at the atomic and molecular levels67,68,69. This theoretical framework enables the detailed analysis of electron density distributions and the topological features of chemical bonds and presents an essential understanding of adsorption. Therefore, by conducting QTAIM analysis, we have defined the nature and strength of intermolecular and intramolecular interactions within the systems resulting in the clarification of the mechanisms underlying the adsorption process. Herein, crucial insights were gained by identifying critical points in the electron density distribution, such as bond critical points (BCPs), which characterize the regions of significant electron density accumulation between atoms68. These BCPs provide valuable information about the nature of chemical bonding, including covalent, ionic, or van der Waals interactions, thus, providing insights into the stability and geometry of adsorbate-adsorbent systems. Furthermore, this analysis facilitates the investigation of charge transfer phenomena and the redistribution of electron density upon adsorption. The calculated results presented in Table S1 and Figure S1 demonstrated a positive value of the Laplacian of charge density, as observed in the intermolecular and intramolecular regions. This indicates a decrease in the values of charge density between the modified system and gas molecules, thus presenting a typical characteristic of closed-shell interactions. The Laplacian of charge density value of ∇2 ρ(r) at 85,73, and 62 critical points (CP) of the NO2-N-In-Rh@C60 system was relatively higher (0.4516 a.u, 0.5833 a.u, and 0.3699 a.u) as compared to the other systems. Furthermore, electron localization and delocalization within the systems were investigated through the electronic localization function (ELF), where an ELF value less than zero indicates high delocalization, while a value greater than zero is indicative of localization regions70,71. All the studied systems exhibited delocalization of electrons at most of their critical points (see Table S1). However, the least value of ELF (0.2921 a.u) was observed at the bond S93-N107 and 73 critical points, as well as the In61-N63 bond length for the NO2-N-In-Rh@C60 system, indicating electronic mobilization between the atom of the gas and the modified system. Additionally, more characteristics of the interactions can be accounted for by the interaction between electrons and nuclei of the systems, and the value of potential energy density V(r) can be utilized for this prediction, as higher values indicate significantly strong interactions72. Surprisingly, the V(r) values recorded from some critical points were negative across all the systems, indicating a weak force of interactions. This analysis reveals key findings on adsorbate-adsorbent interactions, enhancing the understanding of their electronic structure and bonding characteristics in sensor materials.

3D plots of the non-covalent interactions of the systems.
Sensor mechanisms
Determining how the newly tailored material can be used in real-time applications, such as in the design of a sensor device, can be achieved through sensing mechanisms. Here, the work function, fraction of electron transfer, back-donation, and conductivity were utilized to determine the material’s suitability for sensing applications.
Change in work function
The percentage change in the work function (%Δϕ) was computed to examine the percentage increase of the work function during the adsorption process. Before adsorption, the newly engineered materials possessed a work function of 4.914 eV, and this was found to increase upon adsorption to 9.299 (89.24%), 8.823 (79.55%), 8.501 (73.00%), and 8.324 eV (69.39%) for NO2-N-In-Rh@C60, NO2-O-In-Rh@C60, NO-N-In-Rh@C60, and NO-O-In-Rh@C60 respectively (see Table 5). However, a slight decrease in work function was observed for NH3-In-Rh@C60 with a work function and percentage increase of 4.329 (-11.91%). Several studies have shown that an increase in the work function of a system is a desirable attribute for performing the required function, and in this case, the said attribute is the adsorbing tendency.
Charge transfer
The natural charge on the adsorbate and adsorbent were computed to investigate the mechanism of charge transfer (Qt). Theoretically, Qt can be calculated as the difference between the charge on the adsorbate and that on the adsorbent, as shown in Eq. (9)73:
$$\:{Q}_{t}={Q}_{adsorbent}-{Q}_{adsorbate}$$
(9)
The charge transfer from the adsorbent to the adsorbate was observed for all the systems, as indicated by their positive Qt values (see Table 5). This implies that the transfer of electrons has occured, confirming the interaction between the gas molecules and the In-Rh@C60 system.
Electronic conductivity
Mathematically, the electrical conductivity can be related to the energy gap via Eq. (10) as follows:
$$\:{\upsigma\:}=\text{A}{T}^{2/3}{e}^{\left({E}_{g}/2KT\right)}$$
(10)
where electrical conductivity (σ), Boltzmann constant (k), temperature (T), energy gap (Eg), and pre-exponential factor (A) are denoted accordingly. Based on this equation, a high value of Eg implies high conductivity74. The energy gap values are within a relatively small range of 0.705 to 1.537 eV, showing a considerable and acceptable level of conductivity, which is in agreement with previously reported findings75,76. In most cases, the energy gap is reduced upon adsorption, and this has been expressed in the percentage change in the energy gap summarized in Table 5. The highest change in the energy gap of -0.515 (51.51%) and -0.389 (38.93%) is associated with the adsorption of NO gas at the O- and N-sites, respectively. These results reveal the contribution of the modified In-Rh@C60 fullerene system in demonstrating desirable adsorption properties while reducing the energy gap upon adsorption.
The FET and Back-donation mechanisms
The fraction of electron transfer (FET) can be obtained when electrons move from the tailored system to the gases. Similarly, the mobility of electrons back from the gases to the modified In-Rh@C60 system can be illustrated using the electrical back donation. A material suitable for sensing gases often has a negative back donation (ΔE < 0) with positive global hardness (η > 0). Equations (11) and (12) present the mathematical relationships used to calculate the FET and back-donation based on the Pearson theory77.
$$\:\varDelta\:N=\frac{{{\upchi\:}}_{\varvec{i}\varvec{s}\varvec{o}\varvec{l}\varvec{a}\varvec{t}\varvec{e}\varvec{d}}-\:{{\upchi\:}}_{\varvec{s}\varvec{y}\varvec{s}\varvec{t}\varvec{e}\varvec{m}}}{2\:\left({\eta\:}_{isolated\:}-\:{\eta\:}_{system}\right)}$$
(11)
$$\:{\varDelta\:E}_{Back\:donation}=-\frac{\mathbf{\eta}}{4}$$
(12)
In all cases, the back donation values are negative (ΔE < 0), with the highest negative values of -0.1923, -0.1443, and -0.1110 corresponding to NO-O-In-Rh@C60, NO-N-In-Rh@C60, and NO2-N-In-Rh@C60. The least negative ΔE value of -0.08800 is attributed to NO2-O-In-Rh@C60. These results correlate with those obtained in the adsorption energy and are consistent with the observed order of adsorption strength.